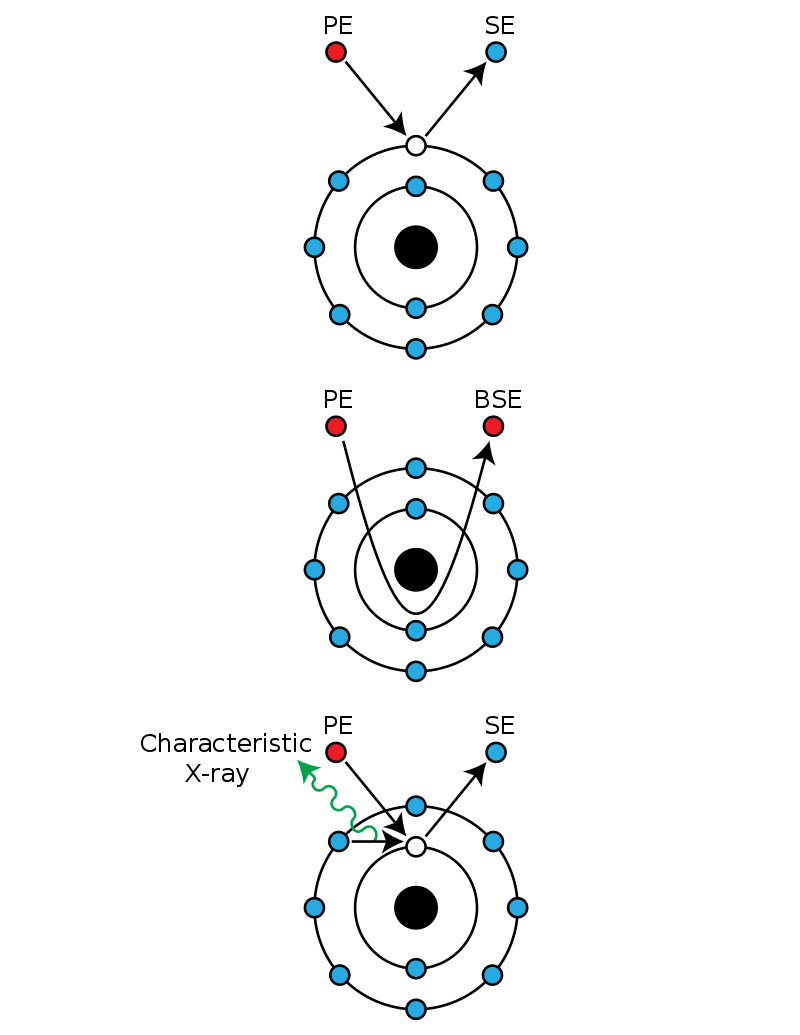
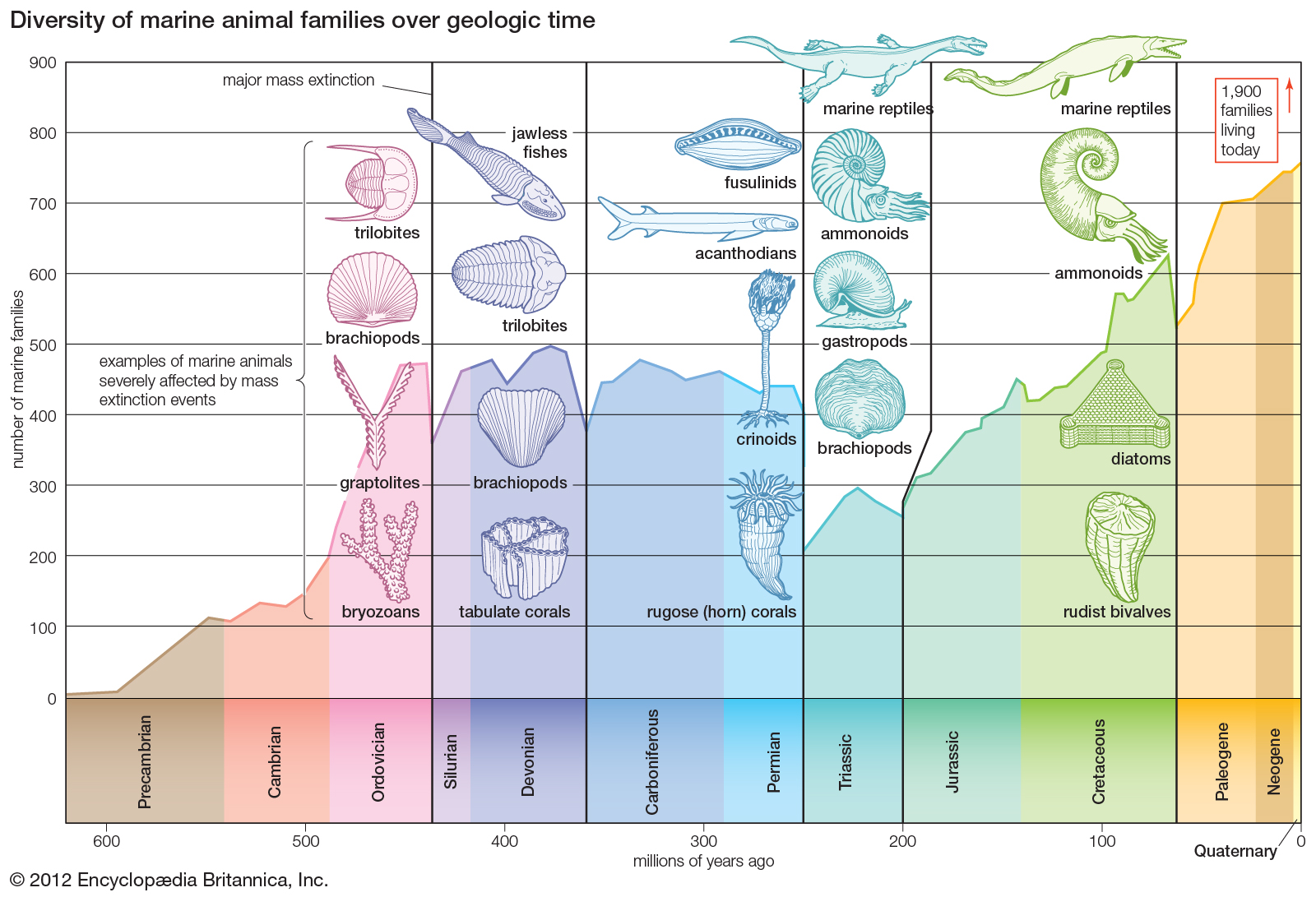
X-ray crystallography (XRC) is the experimental science determining the atomic and molecular structure of a crystal, in which the crystalline structure causes a beam of incident X-rays to diffract into many specific directions. By measuring the angles and intensities of these diffracted beams, a crystallographer can produce a three-dimensional picture of the density of electrons within the crystal. From this electron density, the mean positions of the atoms in the crystal can be determined, as well as their chemical bonds, their crystallographic disorder, and various other information.
Since many materials can form crystals — such as salts, metals, minerals, semiconductors, as well as various inorganic, organic, and biological molecules — X-ray crystallography has been fundamental in the development of many scientific fields. In its first decades of use, this method determined the size of atoms, the lengths and types of chemical bonds, and the atomic-scale differences among various materials, especially minerals and alloys. The method also revealed the structure and function of many biological molecules, including vitamins, drugs, proteins and nucleic acids such as DNA. X-ray crystallography is still the primary method for characterizing the atomic structure of new materials and in discerning materials that appear similar by other experiments. X-ray crystal structures can also account for unusual electronic or elastic properties of a material, shed light on chemical interactions and processes, or serve as the basis for designing pharmaceuticals against diseases.
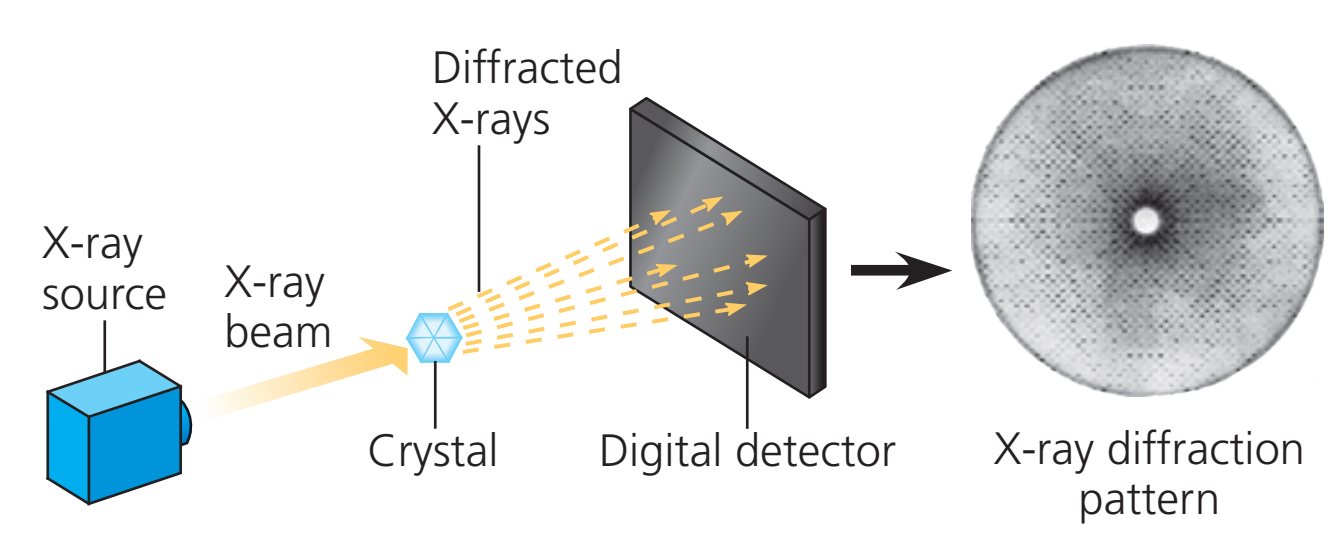
In a single-crystal X-ray diffraction measurement, a crystal is mounted on a goniometer. The goniometer is used to position the crystal at selected orientations. The crystal is illuminated with a finely focused monochromatic beam of X-rays, producing a diffraction pattern of regularly spaced spots known as reflections. The two-dimensional images taken at different orientations are converted into a three-dimensional model of the density of electrons within the crystal using the mathematical method of Fourier transforms, combined with chemical data known for the sample. Poor resolution (fuzziness) or even errors may result if the crystals are too small, or not uniform enough in their internal makeup.
X-ray crystallography is related to several other methods for determining atomic structures. Similar diffraction patterns can be produced by scattering electrons or neutrons, which are likewise interpreted by Fourier transformation. If single crystals of sufficient size cannot be obtained, various other X-ray methods can be applied to obtain less detailed information; such methods include fiber diffraction, powder diffraction and (if the sample is not crystallized) small-angle X-ray scattering (SAXS). If the material under investigation is only available in the form of nanocrystalline powders or suffers from poor crystallinity, the methods of electron crystallography can be applied for determining the atomic structure.
For all above mentioned X-ray diffraction methods, the scattering is elastic; the scattered X-rays have the same wavelength as the incoming X-ray. By contrast, inelastic X-ray scattering methods are useful in studying excitations of the sample such as plasmons, crystal-field and orbital excitations, magnons, and phonons, rather than the distribution of its atoms.
Workflow for solving the structure of a molecule by X-ray crystallography. |
|

A powder x-ray diffractometer in motion. |
|
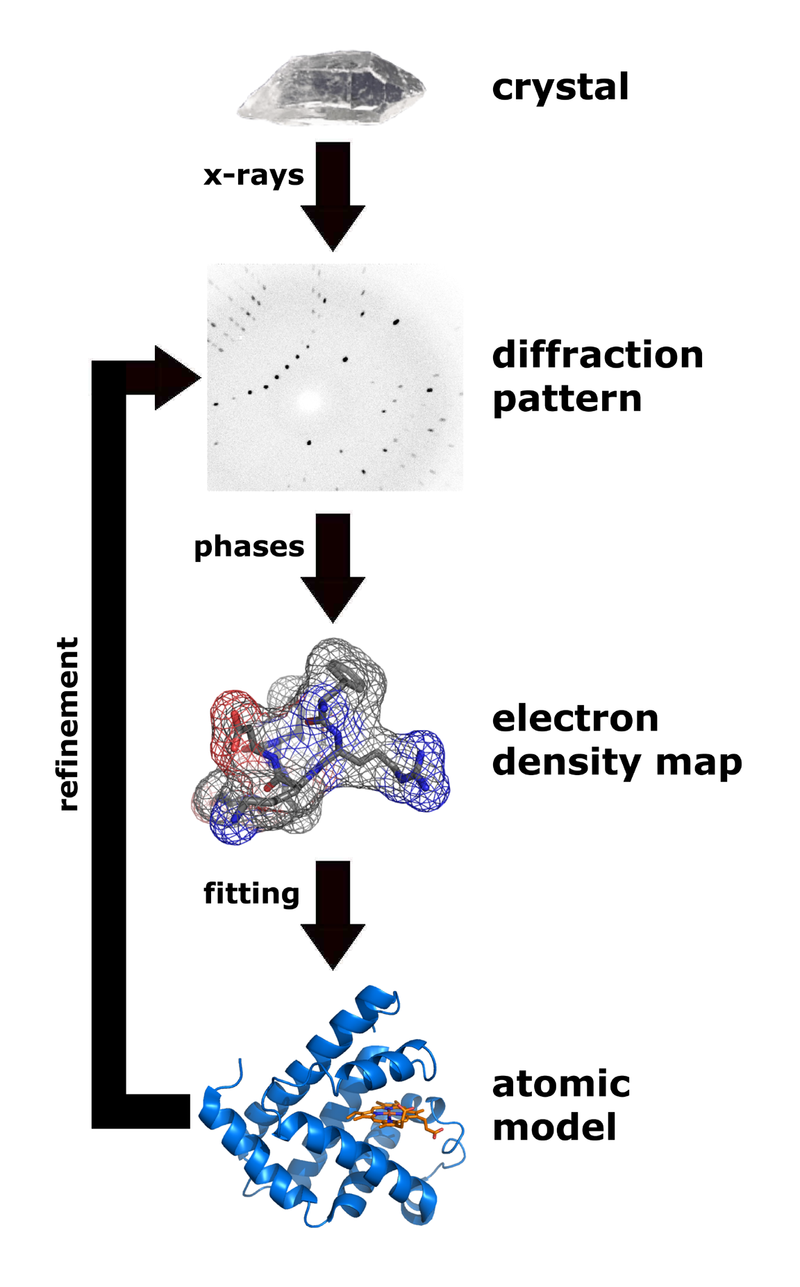
X-ray crystallography shows the arrangement of water molecules in ice, revealing the hydrogen bonds (1) that hold the solid together. Few other methods can determine the structure of matter with such precision (resolution). |
|
Biological macromolecular crystallography
Crystal structures of proteins (which are irregular and hundreds of times larger than cholesterol) began to be solved in the late 1950s, beginning with the structure of sperm whale myoglobin by Sir John Cowdery Kendrew, for which he shared the Nobel Prize in Chemistry with Max Perutz in 1962. Since that success, over 130,000 X-ray crystal structures of proteins, nucleic acids and other biological molecules have been determined. The nearest competing method in number of structures analyzed is nuclear magnetic resonance (NMR) spectroscopy, which has resolved less than one tenth as many. Crystallography can solve structures of arbitrarily large molecules, whereas solution-state NMR is restricted to relatively small ones (less than 70 kDa). X-ray crystallography is used routinely to determine how a pharmaceutical drug interacts with its protein target and what changes might improve it. However, intrinsic membrane proteins remain challenging to crystallize because they require detergents or other denaturants to solubilize them in isolation, and such detergents often interfere with crystallization. Membrane proteins are a large component of the genome, and include many proteins of great physiological importance, such as ion channels and receptors. Helium cryogenics are used to prevent radiation damage in protein crystals.
On the other end of the size scale, even relatively small molecules may pose challenges for the resolving power of X-ray crystallography. The structure assigned in 1991 to the antibiotic isolated from a marine organism, diazonamide A (C40H34Cl2N6O6, molar mass 765.65 g/mol), proved to be incorrect by the classical proof of structure: a synthetic sample was not identical to the natural product. The mistake was attributed to the inability of X-ray crystallography to distinguish between the correct -OH / -NH and the interchanged -NH2 / -O- groups in the incorrect structure. With advances in instrumentation, however, similar groups can be distinguished using modern single-crystal X-ray diffractometers.
Despite being an invaluable tool in structural biology, protein crystallography carries some inherent problems in its methodology that hinder data interpretation. The crystal lattice, which is formed during the crystallization process, contains numerous units of the purified protein, which are densely and symmetrically packed in the crystal. When looking for a previously unknown protein, figuring out its shape and boundaries within the crystal lattice can be challenging. Proteins are usually composed of smaller subunits, and the task of distinguishing between the subunits and identifying the actual protein, can be challenging even for the experienced crystallographers. The non-biological interfaces that occur during crystallization are known as crystal-packing contacts (or simply, crystal contacts) and cannot be distinguished by crystallographic means. When a new protein structure is solved by X-ray crystallography and deposited in the Protein Data Bank, its authors are requested to specify the “biological assembly” which would constitute the functional, biologically-relevant protein. However, errors, missing data and inaccurate annotations during the submission of the data, give rise to obscure structures and compromise the reliability of the database. The error rate in the case of faulty annotations alone, has been reported to be upwards of 6,6% or approximately 15%, arguably a non-trivial size considering the number of deposited structures. This “interface classification problem” is typically tackled by computational approaches and has become a recognized subject in structural bioinformatics. |







.JPG)

.png)