What is a polymerase chain reaction? Polymerase chain reaction is a technique used to massively multiply DNA, so that it can be analyzed using other techniques. It requires a thermal cycler, filled with the DNA sample to be multiplied, thermostable Taq polymerase, primers with which we select what gets multiplied, and free nucleotides of all 4 types - A, T, C and G. The first step is denaturation, then there’s annealing, and finally extension.
📹 PCR — Polymerase Chain Reaction / Henriks World (VİDEO)
📹 PCR — Polymerase Chain Reaction — Henriks World (LINK)
Literature:
Saiki, R. K., Scharf, S., Faloona, F., Mullis, K., Hoorn, G. T., & Arnheim, N. (1985). Polymerase chain reaction. Science, 230, 1350-1354.
This PCR introduction will demonstrate that PCR is a fundamental technique used to amplify fragments of DNA, frequently using the Taq polymerase to facilitate the amplification during the thermal cycling process.
There are many variations of the PCR process including RT PCR for reverse transcription from an RNA template, qPCR for quantitative real time PCR to quantify the amplification during the chain reaction, and many more techniques to optimize the process.
In this PCR video animation, we will give you an overview of the polymerase chain reaction steps as well as the role Taq DNA Polymerase plays in the PCR process. You will learn about PCR applications and factors important in the design of your PCR experiment.
How do PCR DNA polymerases work? ➜ DNA polymerases are essential for the All forms of PCR including, real time quantitative PCR (qPCR), RT PCR, qRTPCR and reverse transcription, multiplex PCR, touch-down PCR and more. Polymerases are the responsible enzyme for nucleotide assembly and extension thus, essential to the assembly of new DNA molecules. However, there are a number of additional PCR components that also need to be taken into consideration. These included the thermal cycle conditions, fidelity, processivity and buffer components for the polymerase chain reaction. These DNA polymerases are indispensable to cell divisions as they duplicate the genetic information that would be passed to the next generation of daughter cells. Throughout the years, scientists have utilized different variants of DNA polymerases for different applications and prove to be essential for modern molecular biology.
➜ Real-Time PCR or quantitative PCR (qPCR) is a PCR-based technique that is able to simultaneously amplify and detect changes in the amplicon concentration. Real-time PCR collects data during PCR amplification by utilizing fluorescence signals emitted by either special probes or DNA binding dyes.
**Please note that the labels of 5’ and 3’ are flipped at 4:35 – 5:22. Usually R (reporter) is at the 5' end and Q (quencher) is at the 3' end, and thus the top strand bound by the Taqman Probe should be labelled as 3’ on the left, and 5’ on the right. Vice versa for the bottom strand: this should be labelled as 5’ on the left, and 3’ on the right. Please refer to Figure 3 of our knowledge base: https://bit.ly/2Xwdybj. We apologize for the confusion and thank our youtube community for pointing out the error.
📹 The principle of PCR-Polymerase Chain Reaction, a full and easy explanation / Biomedical and Biological Sciences (VİDEO)
📹 The principle of PCR-Polymerase Chain Reaction, a full and easy explanation / Biomedical and Biological Sciences (LINK)
This video explains completely and easily PCR, the technique, the principle and the protocol.
We live in a moment where genetics is helping us understand more and more of the world around us, from untangling evolutionary histories to more precisely diagnosing and treating disease. But to do all of this, we need to be able to find and examine specific pieces of DNA. One of the fundamental tools that we use to do this is called PCR, or Polymerase Chain Reaction.
Join The Amoeba Sisters as they explain the biotechnology PCR. This video goes into the basics of how PCR works as well as two examples of its potential use.
Table of Contents:
00:00 Intro
1:34 How does PCR work?
4:31 Why use PCR?
5:10 rRT-PCR testing for SARS-CoV-2 (virus that causes COVID-19)
📹 Real Time PCR — Basic simple animation — Part 1 Intro /
MrSimpleScience (VİDEO)
📹 Real Time PCR — Part 1 Intro /
MrSimpleScience (LINK)
This is a basic video about quantitative/ Real Time PCR. This is just an introduction. More videos will follow. Watch the other videos as well.
📹 qPCR Probe Animation Video / LGC Biosearch Technologies (VİDEO)
📹 qPCR Probe Animation Video / LGC Biosearch Technologies (LINK)
This video showcases these different real-time PCR probe and primer formats: Dual-labeled BHQ probes, BHQplus probes, Molecular Beacons, and Scorpions Primers.
I make animations in biology with PowerPoint, this animation video is about real-time PCR (qPCR). A method by which the amount of the PCR product can be determined, in real-time.
📹📹📹 PCR 2 (VİDEO)
📹 The Evolution of PCR / RocheDiagnosticsUSA (VİDEO)
📹 The Evolution of PCR / RocheDiagnosticsUSA (LINK)
The discovery of Polymerase Chain Reaction (PCR) technique, by Kary B. Mullis, allowed scientists to generate millions of copies of DNA. In 1991, Roche bought the rights to PCR from Cetus and invested in refining the science for use in molecular diagnostics to detect diseases. Launching the first FDA-approved PCR test in 1993, Roche Molecular Diagnostics has not only defined and refined PCR, but it has remained the clear leader of this technology.
This video lecture explains in detail the Basics of Polymerase Chain reaction. Besides the details of this process, we will also understand what do we mean by a long chain and short chain in PCR.
Leicester University geneticist Alec Jeffreys developed a technique called DNA fingerprinting in 1985. It allows DNA samples from different people to be compared to look for similarities and differences. It is useful for solving crimes and can also confirm if people are related to each other, like in paternity testing. Any two people in the world have 99.9% of their DNA the same, so this process analyses the differences in the remaining 0.1%.
This modern technology is called DNA profiling.
📹 PCR Primer Design / Thermo Fisher Scientific (VİDEO)
📹 PCR Primer Design / Thermo Fisher Scientific (LINK)
Tips for PCR Primer Design.
📹 Coronavirus Test — Real time RT-PCR / Biology with Animations (VİDEO)
📹 Coronavirus Test — Real time RT-PCR / Biology with Animations (LINK)
I make animations in biology with PowerPoint, this animation video is about the standard coronavirus test, real time RT-PCR method, which is a laboratory technique combining reverse transcription of RNA into complementary DNA, and amplification of specific DNA targets using polymerase chain reaction (PCR).
Polimeraz zincir tepkimesi (PCR) belirli bir DNA bölümünün yineleyen döngüler yoluyla milyonlarca ve milyarlarca eşlemini yapmak için yaygın olarak kullanılan bir yöntemdir.
PCR için DNA polimeraz enzimi kullanılır.
DNA polimeraz tüm dirimli varlıklarda bulunur ve DNA eşlenimi sırasında hücreler bölünmeden önce DNAnın bir eşini yapar.
“Zincir tepkime” bu bağlamda önceki döngünün ürününün sonraki döngü için başlangıç gereci olarak alınmasını ve böylelikle katlanarak çoğalmasının olanaklı kılınmasını anlatır (1, 2, 4, 8, 16, 32 ...).
PCR 1983’te Amerikalı biokimyacı Kary Mullis tarafından Cetus Corporation’da geliştirildi (Mullis’e buluşundan ötürü 1993 Nobel kimya ödülü verildi).
PCR biolojik ve tıbbi laboratuarlarda örneğin kalıtımsal hastalıkları ve viral enfeksiyonları saptama, genetik parmak izi yaratma ve denetleme, genleri klonlama ve ebeveyn doğrulama işlemlerinde kullanılır.
PCR moleküler biolojinin en önemli yöntemlerinden biridir ve bu alanda birçok bilimsel ilerlemeyi (örneğin insan genomu projesi bağlamında) olanaklı kılan yöntemdir.
Kary B. Mullis was an LSD-dropping, climate-change-denying, astrology-believing, board surfing, Nobel Prize-winning chemist who was both widely respected and equally criticized for his controversial views. (L)
Kary Mullis’in yönteminde DNA polimeraz işlem sırasında tekil bir DNA teline bağlanır ve ona tümleyici olan bir telin bireşimi için kısa, tümleyici bir oligonükleotid (primer) kullanır.
Çift-telli DNA ilkin 96 ° C’ye ısıtılarak iki tekil tele ayrılır.
Bu sıcaklıkta başlangıçta kullanılan E. coli DNA polimeraz I enzimi bozulur ve her ısıtmadan sonra yeniden eklenmesi gerekir.
Yeterli artışa ulaşmak için, bu iş sırası arka arkaya düzinelerce kez yinelenir.
Mullis’in başlangıçtaki yöntemi çok yetersiz idi ve büyük miktarlarda DNA polimeraz ve sürekli dikkat gerektiriyordu.
Yaklaşık 100° C’de bile polimeraz etkinliklerini sürdüren ve denatüre olmayan ısıya-dayanıklı DNA polimerazların kullanımı PCR uygulayımında önemli ilerlemelere götürdü.
Isıya-dayanıklı ilk DNA polimerazlardan biri çok sıcak kaynaklarda yaşayan termofilik bir bakteri olan Thermus aquaticustan elde edildi (Taq polimeraz).
Isıya-dayanıklı PCR süreci önemli ölçüde yalınlaştı ve otomatikleşti.
Mullis 1980’lerde California bioteknik şirketi Cetus’ta çalışıyordu ve 10.000 dolarlık bir ikramiye ile ödüllendirildi.
Yıllar sonra Cetus PCR yönteminin ve Taq DNA polimerazın haklarını Roche’ye 300 milyon dolara sattı.
PCR teknolojisi için ABD patentleri 2005’te sona erdi.
🛑 ISIL DÖNGÜ
Iısıl döngü (Thermal cycle)
Yüksek işlem hacımlı modern bir ısıl çevrimci (PCR makinesi ya da DNA arttırıcı).
PCR yöntemlerinin büyük çoğunluğu genellikle 25-50 kez yinelenen bir sıcaklık değişimleri dizisinden oluşan ısıl döngü (thermal cycle) üzerine dayanır.
Isıl döngü işleme katılan tepkenleri (reactant) yineleyen ısıtma ve soğutma döngülerine sokarak değişik ısı-bağımlı tepkimelere girmelerini sağlar (örneğin DNA eritme ve enzim-güdümlü DNA eşlenimi).
PCR işlemi etkinleştiren iki ana tepkin (reagent) kullanır — (1) primerler (hedef DNA bölgesine tümleyici bir dizi olan ve oligonükleotidler olarak bilinen tek-telli kısa DNA fragmanları), ve (2) bir DNA polimeraz.
Bu döngüler üç evreden oluşur:
Yaklaşık 95° C’de, birincisi DNAnın çift-telli zincirinin ayrılmasına izin verir.
Yaklaşık 50-60° C’de, ikincisi primerlerin DNA kalıp ile bağlanmasına izin verir.
Yaklaşık 68-72° C’de, üçüncüsü DNA polimeraz tarafından yerine getirilen polimerizasyonu kolaylaştırır.
Polimeraz zincir tepkimesinin üç-adım süreci.
PCR’nin ilk adımında, DNA çift sarmalının iki teli nükleik asit denatürasyonu (ya da erimesi) denilen bir süreçte yüksek ısıda fiziksel olarak ayrılır.
PCR’nin ikinci adımında, sıcaklık düşürülür ve primerler tümleyici DNA dizilerine bağlanır.
Bu aşamada iki DNA teli DNA polimeraz için kalıplar olur ve enzim serbest DNA için yapı-taşları olan nükleotidlerden yeni bir DNA teli bireştirir.
PCR ilerlerken, yaratılan DNAnın kendisi eşlenim için kalıp olarak kullanılır ve tepkimeler zincirinde kökensel DNA kalıbı katlanarak çoğalır.
Hemen hemen tüm PCR uygulamaları ısıya-dayanıklı bir DNA polimeraz kullanır (ilkin termofilik bakteri Thermus aquaticustan yalıtılan Taq polimeraz gibi).
Eğer kullanılan polimeraz ısıya-dayanıksız ise, denatürasyon adımının yüksek sıcaklıklarında denatüre olacaktır.
🛑 İLKELER, YORDAM, EVRELER
🛑 İLKELER
İLKELER
PCR yöntemlerinin çoğu 0,1 ve 10 kilo baz çift (kbç) uzunluktaki DNA fragmanlarını arttırır (kimi uygulayımlar 40 kbç boyundaki fragmanların arttırılmasına izin verir).
Arttırılmış ürün miktarı tepkime için eldeki tözler tarafından belirlenir.
Temel bir PCR düzeni çeşitli bileşenler ve tepkinler gerektirir:
Arttırılacak DNA hedef bölgesini kapsayan bir DNA kalıbı.
DNA polimeraz (yeni DNA tellerini polimerize eden bir enzim; ısı-dayanıklı Taq polimeraz özellikle yaygındır).
DNA hedefin anlam ve karşı-anlam tellerinin her birinin 3' ucuna tümleyici olan iki DNA primeri (DNA polimeraz ancak DNAnın çift telli bir bölgesine bağlanabilir ve oradan uzatma yapabilir; primerler olmaksızın polimerazın bağlanabileceği çift-telli bir başlama sitesi yoktur); DNA hedef bölgesine tümleyici olan özgül primerler önceden seçilir ve sık sık laboratuarda önceden yapılır ya da sunumculardan satın alınır.
DNAnın yapı taşları olan deoxinükleosid trifosfatlar, ya da dNTPler (kimi zaman “deoxinükleotid trifosfat” denir; trifosfat kapsayan nükleotidlerdir).
Tampon çözelti (DNA polimerazın optimum etkinliği ve kararlılığı için uygun bir kimyasal çevre sağlar).
Bivalent kationlar; tipik olarak magnezium (Mg) ya da manganez (Mn) ionları; Mg2+ en yaygın olanıdır, ama Mn2+PCR-dolaylı DNA mutagenesisi için kullanılabilir.
Tepkime genellikle bir ısıl çevrimcide küçük tepkime tüplerinde (0.2–0.5 mL hacımlar) 10–200 μL büyüklüğünde bir hacımda yerine getirilir.
Isıl çevrimci tepkime tüplerini tepkimenin her bir adımının gerektirdiği sıcaklığı elde etmek üzere ısıtır ve soğutur.
🛑 YORDAM
YORDAM
Tipik olarak, PCR ısıl döngüler adı verilen 20-40 kadar yineleyen sıcaklık değişiminden oluşur.
Her bir döngü genellikle iki ya da üç ayrı sıcaklık adımından oluşur.
Döngü süreci sık sık çok yüksek bir derecedeki (>90 °C) tekil bir sıcaklık adımı tarafından öncelenir.
Her bir döngüde kullanılan sıcaklıklar ve uygulanma süreleri bir dizi parametre üzerine bağımlıdır (DNA bireşimi için kullanılan enzim; tepkimedeki bivalent ionların ve dNTPlerin yoğunluğu; ve primerlerin erime derecesi.
PCR yöntemlerinin çoğuna ortak tekil adımlar şöyledir:
Başlatma: Bu adım yalnızca sıcak başlangıç yapan PCR tarafından ısı etkinleştirmesi isteyen DNA polimerazlar için gereklidir; eğer ısıya aşırı dayanıklı polimerazlar kullanılırsa, tepkime odasının 94-96° C ya da 98° C düzeyine ısıtılmasından oluşur.
Denatürasyon: Bu adım ilk düzenli çevrim olayıdır ve tepkime odasının 20-30 saniye kadar 94-98° C düzeyine ısıtılmasından oluşur; bu işlem tümleyici bazlar arasındaki hidrojen bağlarının koparılması yoluyla çift-telli DNAnın erimesine ya da denatürasyonuna neden olur ve iki tek-telli DNA molekülü ortaya çıkar.
Tavlama ya da sertleştirme: Sonraki adımda tepkime sıcaklığı 20-40 sn kadar 50-65° C düzeyine düşürülür ve tek-telli DNA kalıplarından her birinin primerlerinin tavlanması sağlanır; tepkime karışımında genellikle hedef bölgeyi kapsayan iki tek-telli tümleyicinin her biri için bir adet olmak üzere iki ayrı primer kapsanır; primerler kendileri tekil-telli dizilerdir, ama hedef bölgenin uzunluğundan çok daha kısadırlar ve yalnızca her bir telin 3' ucunda çok kısa dizileri tümlerler. (Tavlama adımı için uygun bir sıcaklık belirlemek çok önemlidir, çünkü etkerlik ve özgüllük tavlama sıcaklığı tarafından güçlü olarak etkilenir.)
Uzatma. Bu adımdaki sıcaklı kullanılan DNA polimeraza bağımlıdır; bu adımda DNA polimeraz DNA kalıp teline tümleyici olan yeni bir DNA teli bireştirir ve bunu tepkime karışımından 5'—3' yönünde kalıba tümleyici olan serbest dNTPler ekleyerek ve dNTPlerin 5'-fosfat grubunu doğmakta olan (uzayan) DNA telinin ucundaki 3'-hidroksi grubu ile yoğuşturarak yapar. DNA polimerazların çoğu dakikada bin kadar bazı polimerize eder. Optimal koşullar altında, her bir uzatma adımında DNA hedef dizilerinin sayısı iki katına çıkar. Her bir ardışık döngü ile, kökensel kalıp telleri artı yeni yaratılan teller sonraki uzatma aşaması için kalıp teller olur ve özgül DNA hedef bölgesinin katlanarak (geometrik, eksponensiyel) artışı sağlanır.
🛑 EVRELER
EVRELER
Bütün PRC süreci tepkimenin ilerlemesine göre üç evreye bölünebilir.
Katlamalı arttırma: Her döngüde, ürünün miktarı iki katına çıkar (%100 tepkime etkerliği varsayılarak). 30 döngüden sonra, tek bir DNA eşlemi 1.000.000.000 (bir milyar) eşleme arttırılabilir.
Düzleştirme evresi: DNA polimeraz etkinlik yitirdikçe ve dNTPler ve primerler gibi tepkinlerin tüketimi daha sınırlı olmalarına yol açtıkça tepkime yavaşlar.
Düzlek: Tepkinlerin ve enzimin tükenmesinden ötürü daha fazla ürün birikimi olmaz.
Bir DNA dizisi eğer proteine çevrilen iletmen RNA eşleminin dizisi ile aynı yapıda ise bir “anlam” dizisidir.
Karşıt telin dizisi “karşı-anlam” dizisidir.
Hem anlam hem de karşı-anlam dizileri aynı DNA telinin değişik parçaları üzerinde olabilir (e.d. her iki tel de hem anlam hem de karşı-anlam dizileri kapsayabilir).
Karşı-anlam RNA dizilerinin işlevi bütünüyle açık değildir, ama RNA-RNA baz çiftlenmesi yoluyla gen anlatımını düzenliyor olabilirler.
Kimi DNA dizileri çakışan genler taşıdıkları için, anlam ve karşı-anlam arasındaki ayrım bulanıktır.
📹 Difference between Sense Strand and Antisense Strand of DNA / biologyexams4u (VİDEO)
📹 Difference between Sense Strand and Antisense Strand of DNA / biologyexams4u (LINK)
This video explains
Plus strand vs minus strand, coding vs non-coding strand, template vs non-template strand of DNA
sense strand vs antisense strand of DNA
DNA polimeraz çifte-sarmal DNAnın çevresine sarılıyor. —
DNA polimerazın yapısı avuç içi, parmaklar ve baş parmak ile bir sağ ele benzetilir (avunç iç etkin sitedir..
DNA polimeraz — nükleotid baz eşlem ve onarımı.
DNA eşlenimine özsel olan DNA polimeraz DNA moleküllerinin nükleosid trifosfatlardan bireşimini katalize eden enzimler grubuna aittir (ayrıca DNA bakım ve eşlenimine de katılır).
Eşlenim süreci sırasında DNA polimeraz varolan DNA telini “okur” ve varolan iki telin eşlemleri olan yeni iki telin yaratılmasına aracılık eder.
Bu enzimlerin katalize ettikleri kimyasal tepkime:
DNA polimeraz nükleotidleri birer birer DNA telinin 3' (üç asal) ucuna ekler.
Eşlemenin yer alabilmesi için, helikaz enzimi DNA molekülünün iki telini ayırır ve bunu yapmak için nükleotid bazlar arasındaki hidrojen bağlarını kırar.
İki tekil DNA teli yukarıda verilen tepkimedeki eşlenim için kalıplar olarak kullanılır.
DNA polimeraz eski tel boyunca 3'–5' yönünde ilerler ve 5'–3' yönünde olan yeni bir tel yaratır.
Oligonükleotid sentezi belirli kimyasal yapıları (dizi) ile nükleik asit fragmanlarının kimyasal bireşimidir.
Enzimler DNA ve RNAyı yalnızca 5'—3' yönünde birleştirirken, kimyasal oligonükleotid bireşimi bu sınırlamayı tanımaz ve sık sık karşıt 3'—5' yönünde yerine getirilir.
Doğal yaşamda kısıtlama enziminin işlevi bakteriyi bakteriofaj denilen özgül viruslara karşı savunmaktır.
Bu viruslar bir bakteriyel plazmide viral RNA ya da DNA aşılayarak ve orada eşlemleme yaparak saldırırlar.
Eğer bir prokaryot hücrenin içerisinde viral RNA ya da DNA bulunacak olursa, o hücre çoğunlukla yabancı genetik diziyi keserek eşlemlemeyi durdurabilir.
Bakteri. Dairesel plazmid sağ altta görülüyor.
Kısıtlama enzimi (restriktaz) DNAyı moleküllerin içerisinde tanıma siteleri olarak bilinen sitelerde ya da bunların yakınında fragmanlara yaran bir biolojik makromoleküldür.
Kısıtlama enzimi bakteride immün dizgenin parçası olarak işlev görür (bakteri ya da arkhea hücre dışından gelen viral DNAyı keserek zararsızlaştırır).
Kısıtlama enzimleri daha geniş endonükleaz enzim grubunun bir sınıfıdır.
Kısıtlama enzimleri genellikle beş tip olarak sınıflandırılır.
DNAyı kesmek için, tüm kısıtlama enzimleri DNA çift sarmalının her bir şeker-fosfat omurgası (= her bir teli) üzerinde olmak üzere iki kesme yapar.
Kısıtlama enzimleri laboratuarlarda DNA değişkisi için kullanılmaktadır ve moleküler klonlamada önemli bir araçtır.
Bugüne dek, bakteriel plasmidlerden 3000’den fazla kısıtlama enzimi yalıtılmış ve incelenmiştir (bunlardan 600’den fazlası tecimsel ortamdadır).
Her bir kısıtlama enzimi özgül bir viral genetik kod dizisini tanır ve tanıma sitesine yakın ya da uzak yeni, mutasyonlu DNA telini ayırmaya çalışır (bu doğal ayırma düzeneği “kısıtlama enzimi sindirimi” olarak da adlandırılır).
HindIII denilen kısıtlama enzimi DNAyı iki tel üzerindeki değişik noktalarda yararak yapışkan bir uç yaratır.
Kısıtlama enzimleri nükleotidlerin belirli bir dizisini tanır ve DNAda bir çift-telli kesme yapar.
Tanıma dizileri tanıma sitesindeki bazların sayısı ile de sınıflandırılabilir (genellikle 4 ve 8 arasında).
Dizideki bazların sayısı sitenin verili bir genomda hangi sıklık ile görünebileceğini belirler (örneğin bir 4-baz çiftli (bp) dizi kuramsal olarak her 4^4 ya da 256bp için, 6-baz çiftli bir dizi 4^6 ya da 4096bp için bir kez görünecektir).
DNA ve RNA molekülleri dört nükleotidin dilinde yazılıdır (DNA: A, T, G, C; RNA: A, U, G, C); proteinlerin dili 20 amino asidi kapsar.
Yukarıdaki kodon tekerleği DNA kodonlarını amino asitlere çevirmek için kullanılabilir. Kodon dizisindeki ilk harf iç dairede seçildikten sonra, karşılık düşen amino asidi görmek dışa doğru ilerlenir. Örneğin ATG = methionin. (
Image credit: Genome Research Limited) (LINK)
Kodon bir DNA ya da RNA molekülünde bir genetik kod birimidir.
Kodonlar üç DNA ya da RNA bazından oluşur.
Her bir kodon belirli bir amino asidi kodlar (örneğin AAA kodonu lisin için, ve CCC kodonu prolin için kodlama yapar.
Olanaklı 64 kodon vardır.
En az 47 başlangıç kodonu vardır ve bunlardan her biri hücreye protein bireşimine başlama sinyalini verebilir.
Olanaklı 64 kodondan üçü dur kodonlarını yapar.
Üç dur kodonu UAG, UAA ve UGAdır (bu kodonlar çeviri sırasında polipeptid zincirin sonunu belirtir; hiçbir amino asit için kodlama yapmadıkları için anlamsız kodonlar ya da sonlandırma kodonları olarak da bilinirler).
Her bir tRNA bir kodonu bir amino asit ile eşler.
ANTİKODON
Antikodon dizisi tRNA molekülleri üzerinde bulunur, kodonlara tümleyicidir, ve protein bireşimi sırasında tRNAnın taşıdığı amino asidi belirler.
Herhangi bir tRNAnın antikodonu o tRNAya bağlı amino asit için kodlama yapan mRNA kodonuna tam olarak uyar.
Olanaklı 64 antikodondan 61’i protein oluşumu için kodlama yapar; üç antikodon protein bireşimini sonlandırma işlevini yerine getirir.
AUC dur antikodonudur.
Bu grafik büyüyen bir protein zincirini gösterir. Sol aşağıya doğru amino asitler taşıyan tRNAların ribozom karmaşasına girdikleri görülebilir. Eğer herşey yolunda giderse, yalnızca doğru antikodonları olan tRNAlar açıktaki mRNAya başarılı olarak bağlanacak ve böylece yalnızca doğru amino asitler eklenecektir.
tRNAlar mRNAların yönergelerine göre proteinlere katılmak üzere doğru amino asitlerin getirilmesinden sorumludur. tRNAların mRNA üzerindeki kodonlar ile eş-bağı kuran antikodonları onlara bu işlevi yerine getirme iznini verecektir.
Bir iletmen RNA (mRNA) molekülünün parçası üzerinde bir kodonlar dizisi. Her bir kodon genellikle tek bir amino aside karşılık düşen üç nükleotidden oluşur. Nükleotidler A, U, G ve C harfleri ile kısaltılır. Bu molekül mRNAdır ve U (urasil) kullanır. DNA bunun yerine T (thymin) kullanır. Bu mRNA molekülü bir ribozoma bu koda göre bir protein birleştirme yönergesini verecektir.
Moleküler klonlama moleküler biolojide yeniden-bileşimli DNA moleküllerini düzenlemek ve konak örgenlikler içerisinde eşlenimlerini yönetmek için kullanılan deneysel yöntemler kümesini belirten bir kapsama terimidir.
“Klonlama” sözcüğü yöntemin özdeş DNA molekülleri taşıyan bir hücreler nüfusu üretmek için bir molekülün eşleniimini içermesi olgusunu belirtmek için kullanılır.
Moleküler klonlama genellikle iki ayrı örgenlikten DNA dizileri kullanır: (1) Klonlanacak DNAnın kaynağı olan tür; (2) yeniden-bileşimli DNAnın eşlenimi için dirimli konak olarak hizmet edecek tür.
Bakteri ve plasmidlerin kullanımı ile moleküler klonlama çizgesi.
Nükleazlar aşağı yukarı her yerde bulunur; primerler, kalıp DNA ve PCR ürünleri ile birlikte DNAyı hızla bozundururlar.
Nükleaz nükleik asitlerin nükleotidleri arasındaki fosfodiester bağları koparabilen bir enzimdir.
Kısıtlama enzimi (endonükleaz) HindIII’ün bir çift-telli DNA molekülünü geçerli bir kısıtlama sitesinde (5'–A|AGCTT–3') yarma işlemi.
Nükleazlar hedef moleküllerinde tek ve çift telli kopmalar yapar.
Nükleazlar DNA onarımının birçok yanında özsel düzenektir.
Belli nükleazlarda bozukluklar genetik kararsızlığa ve immün yetersizliğe neden olabilir.
Nükleazlar moleküler klonlamada da yaygın olarak kullanılır.
Nükleazlar etkinlik yerine göre başlıca iki sınıfa ayrılır: (1) Exonükleazlar nükleik asitleri uçlardan sindirir; (2) Endonükleazlar hedef moleküllerin ortasındaki bölgeler üzerinde etkindir.
Nükleazlar daha öte alt kategorilere ayrılır: (1) DNA üzerinde etkide bulunan deoxiribonükleazlar; ve (2) RNA üzerinde etkide bulunan ribonükleazlar.
Primer DNA parçaları örnek DNAya bağlanır ve polimeraza bir başlangıç noktası verir.
Primer tüm dirimli örgenliklerde DNA bireşiminin başlatılmasında kullanılan kısa bir tek-telli nükleik asittir.
Primerler arasındaki DNA dizisi PCR tarafından arttırılan (amplified) parçadır ve amplikon ya da PCR ürünü olarak değinilir.
DNA eşleminden sorumlu olan DNA polimeraz enzimleri nükleotidleri varolan bir nükleik asidin ancak 3' ucuna ekleyebilir, ve bu durum DNA polimerazın bir tümleyici tele başlayabilmesi için bir primerin önceden kalıba bağlı olmasını gerektirir.
Dirimli örgenlikler yalnızca RNA primerler kullanır.
Biokimyada ve moleküler biolojide in vitro DNA bireşimini gerektiren laboratuar uygulayımları (DNA dizilendirme ve PCR) genellikle DNA primerler kullanır, çünkü bunlar sıcağa daha dayanıklıdır.
An amplicon sequence template that has been prepared for amplification. The target sequence to be amplified is colored green.
DNA eşlenim çatalı. Etiketli RNA primer üstte.
Standard bir PCR için ileri ve ters primerler.
🛑 TAQ POLİMERAZ
Taq polimeraz
Taq polimeraz yüksek ısıya dayanan bir DNA polimeraz I enzimi, ve Thermus aquaticus yüksek ısıya bayılan bir bakteridir.
Taq polimeraz Thermus aquaticustan elde edilir.
Thermus aquaticus çok sıcak kaynaklarda yaşar ve kapsadığı polimeraz enzimi protein denatüre edici yüksek sıcaklık koşullarına dayanıklıdır.
Yeniden-bileşimli DNA (recombinant DNA, rDNA) molekülleri laboratuarda genetik yeniden-bileşim yöntemleri ile oluşturulan moleküllerdir.
Yöntem birçok kaynaktan genetik gereci biraraya getirerek başka türlü genomda bulunmayacak diziler yaratır.
Değişik kaynaklardan fragmanların yeniden-bileşiminin olanağı DNA moleküllerinin tüm örgenliklerde aynı kimyasal yapıyı paylaşmaları ve yalnızca nükleotid dizilerinde ayrımlar göstermeleridir.
Örneğin bitki DNAsı bakteri DNAsı ile ya da insan DNAsı fungus DNAsı ile birleştirilebilir.
Doğada bulunmayan yapay DNA dizileri de kimyasal DNA bireşimi ile yaratılabilir ve dirimli örgenliklerin yapısına katılabilir ve yeniden-bileşimli yabancı proteinlerin üretimi sağlanabilir.
Yeniden-bileşimli DNA yapımı.
Yabancı bir DNA fragmanının bir plasmid vektöre katılımını gösteren bu örnekte beyaz renk ile belirtilen gen yabancı DNA fragmanının katılması ile etkinliğini yitirir
Gerçek-zamanlı polimeraz zincir tepkimesi ayrıca nicel PCR (qPCR) olarak da bilinir.
qPCR moleküler biolojide PCR üzerine dayalı bir laboratuar uygulayımıdır.
qPCR hedeflenen DNA molekülünün artışını PCR sırasında (gerçek zamanda) gözler, yalnızca sonuç olarak değil.
PCR ürünlerini gerçek zamanda saptamanın iki yaygın yöntemi (1) her çift-telli DNA ile katmanlaşan (baz düzlemleri arasına giren) özgül-olmayan floresan boyalar, ve (2) bir folerasan raporcu ile etiketlenmiş oligonükleotidlerden oluşan diziye-özgü DNA sondaları.
🛑 TERİMLER
TERİMLER
döngü:cycle düzlek:plateu eşlenim:replication ısıl:thermal ısıl çevrimci:thermal cycler (makine) ısıl döngü: thermal cycle ısısever:thermophile ısıya-dayanıklı:thermostable tepken:reactant(tepkimede etken, tepkimeye katılan özdek) tepkin:reagent(tepkimeyi etkin kılan; örneğin çözelti, katalist) tümleyici:complementary
A strip of eight PCR tubes, each containing a 100 μL reaction mixture.
Polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1984 by the AmericanbiochemistKary Mullis at Cetus Corporation. It is fundamental to much of genetic testing including analysis of ancient samples of DNA and identification of infectious agents. Using PCR, copies of very small amounts of DNA sequences are exponentially amplified in a series of cycles of temperature changes. PCR is now a common and often indispensable technique used in medical laboratory and clinical laboratory research for a broad variety of applications including biomedical research and criminal forensics.
The majority of PCR methods rely on thermal cycling. Thermal cycling exposes reactants to repeated cycles of heating and cooling to permit different temperature-dependent reactions – specifically, DNA melting and enzyme-driven DNA replication. PCR employs two main reagents – primers (which are short single strand DNA fragments known as oligonucleotides that are a complementary sequence to the target DNA region) and a DNA polymerase. In the first step of PCR, the two strands of the DNA double helix are physically separated at a high temperature in a process called nucleic acid denaturation. In the second step, the temperature is lowered and the primers bind to the complementary sequences of DNA. The two DNA strands then become templates for DNA polymerase to enzymatically assemble a new DNA strand from free nucleotides, the building blocks of DNA. As PCR progresses, the DNA generated is itself used as a template for replication, setting in motion a chain reaction in which the original DNA template is exponentially amplified.
Almost all PCR applications employ a heat-stable DNA polymerase, such as Taq polymerase, an enzyme originally isolated from the thermophilic bacterium Thermus aquaticus. If the polymerase used was heat-susceptible, it would denature under the high temperatures of the denaturation step. Before the use of Taq polymerase, DNA polymerase had to be manually added every cycle, which was a tedious and costly process.
PCR amplifies a specific region of a DNA strand (the DNA target). Most PCR methods amplify DNA fragments of between 0.1 and 10 kilo base pairs (kbp) in length, although some techniques allow for amplification of fragments up to 40 kbp. The amount of amplified product is determined by the available substrates in the reaction, which becomes limiting as the reaction progresses.
A basic PCR set-up requires several components and reagents, including:
a DNA template that contains the DNA target region to amplify
a DNA polymerase; an enzyme that polymerizes new DNA strands; heat-resistant Taq polymerase is especially common, as it is more likely to remain intact during the high-temperature DNA denaturation process
two DNA primers that are complementary to the 3′ (three prime) ends of each of the sense and anti-sense strands of the DNA target (DNA polymerase can only bind to and elongate from a double-stranded region of DNA; without primers, there is no double-stranded initiation site at which the polymerase can bind); specific primers that are complementary to the DNA target region are selected beforehand, and are often custom-made in a laboratory or purchased from commercial biochemical suppliers
deoxynucleoside triphosphates, or dNTPs (sometimes called "deoxynucleotide triphosphates"; nucleotides containing triphosphate groups), the building blocks from which the DNA polymerase synthesizes a new DNA strand
a buffer solution providing a suitable chemical environment for optimum activity and stability of the DNA polymerase
The reaction is commonly carried out in a volume of 10–200 μL in small reaction tubes (0.2–0.5 mL volumes) in a thermal cycler. The thermal cycler heats and cools the reaction tubes to achieve the temperatures required at each step of the reaction (see below). Many modern thermal cyclers make use of the Peltier effect, which permits both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes permit favorable thermal conductivity to allow for rapid thermal equilibrium. Most thermal cyclers have heated lids to prevent condensation at the top of the reaction tube. Older thermal cyclers lacking a heated lid require a layer of oil on top of the reaction mixture or a ball of wax inside the tube.
Typically, PCR consists of a series of 20–40 repeated temperature changes, called thermal cycles, with each cycle commonly consisting of two or three discrete temperature steps (see figure below). The cycling is often preceded by a single temperature step at a very high temperature (>90 °C (194 °F)), and followed by one hold at the end for final product extension or brief storage. The temperatures used and the length of time they are applied in each cycle depend on a variety of parameters, including the enzyme used for DNA synthesis, the concentration of bivalent ions and dNTPs in the reaction, and the melting temperature (Tm) of the primers. The individual steps common to most PCR methods are as follows:
Initialization: This step is only required for DNA polymerases that require heat activation by hot-start PCR. It consists of heating the reaction chamber to a temperature of 94–96 °C (201–205 °F), or 98 °C (208 °F) if extremely thermostable polymerases are used, which is then held for 1–10 minutes.
Denaturation: This step is the first regular cycling event and consists of heating the reaction chamber to 94–98 °C (201–208 °F) for 20–30 seconds. This causes DNA melting, or denaturation, of the double-stranded DNA template by breaking the hydrogen bonds between complementary bases, yielding two single-stranded DNA molecules.
Annealing: In the next step, the reaction temperature is lowered to 50–65 °C (122–149 °F) for 20–40 seconds, allowing annealing of the primers to each of the single-stranded DNA templates. Two different primers are typically included in the reaction mixture: one for each of the two single-stranded complements containing the target region. The primers are single-stranded sequences themselves, but are much shorter than the length of the target region, complementing only very short sequences at the 3′ end of each strand.
It is critical to determine a proper temperature for the annealing step because efficiency and specificity are strongly affected by the annealing temperature. This temperature must be low enough to allow for hybridization of the primer to the strand, but high enough for the hybridization to be specific, i.e., the primer should bind only to a perfectly complementary part of the strand, and nowhere else. If the temperature is too low, the primer may bind imperfectly. If it is too high, the primer may not bind at all. A typical annealing temperature is about 3–5 °C below the Tm of the primers used. Stable hydrogen bonds between complementary bases are formed only when the primer sequence very closely matches the template sequence. During this step, the polymerase binds to the primer-template hybrid and begins DNA formation.
Extension/elongation: The temperature at this step depends on the DNA polymerase used; the optimum activity temperature for the thermostable DNA polymerase of Taq polymerase is approximately 75–80 °C (167–176 °F), though a temperature of 72 °C (162 °F) is commonly used with this enzyme. In this step, the DNA polymerase synthesizes a new DNA strand complementary to the DNA template strand by adding free dNTPs from the reaction mixture that is complementary to the template in the 5′-to-3′ direction, condensing the 5′-phosphate group of the dNTPs with the 3′-hydroxy group at the end of the nascent (elongating) DNA strand. The precise time required for elongation depends both on the DNA polymerase used and on the length of the DNA target region to amplify. As a rule of thumb, at their optimal temperature, most DNA polymerases polymerize a thousand bases per minute. Under optimal conditions (i.e., if there are no limitations due to limiting substrates or reagents), at each extension/elongation step, the number of DNA target sequences is doubled. With each successive cycle, the original template strands plus all newly generated strands become template strands for the next round of elongation, leading to exponential (geometric) amplification of the specific DNA target region.
The processes of denaturation, annealing and elongation constitute a single cycle. Multiple cycles are required to amplify the DNA target to millions of copies. The formula used to calculate the number of DNA copies formed after a given number of cycles is 2n, where n is the number of cycles. Thus, a reaction set for 30 cycles results in 230, or 1,073,741,824, copies of the original double-stranded DNA target region.
Final elongation: This single step is optional, but is performed at a temperature of 70–74 °C (158–165 °F) (the temperature range required for optimal activity of most polymerases used in PCR) for 5–15 minutes after the last PCR cycle to ensure that any remaining single-stranded DNA is fully elongated.
Final hold: The final step cools the reaction chamber to 4–15 °C (39–59 °F) for an indefinite time, and maybe employed for short-term storage of the PCR products.
Schematic drawing of a complete PCR cycle
To check whether the PCR successfully generated the anticipated DNA target region (also sometimes referred to as the amplimer or amplicon), agarose gel electrophoresis may be employed for size separation of the PCR products. The size of the PCR products is determined by comparison with a DNA ladder, a molecular weight marker which contains DNA fragments of known sizes, which runs on the gel alongside the PCR products.
Ethidium bromide-stained PCR products after gel electrophoresis. Two sets of primers were used to amplify a target sequence from three different tissue samples. No amplification is present in sample #1; DNA bands in sample #2 and #3 indicate successful amplification of the target sequence. The gel also shows a positive control, and a DNA ladder containing DNA fragments of defined length for sizing the bands in the experimental PCRs.
This gel was created and imaged by me. Expression of a specific gene in three separate tissues was tested by two primers. As the gel shows, Tissue #1 lacks that gene, whereas Tissue #2 and #3 possess it. A positive control was used to determine that the PCR conditions were adequate and a 1kb ladder was used to determine the size of the bands. (W)
As with other chemical reactions, the reaction rate and efficiency of PCR are affected by limiting factors. Thus, the entire PCR process can further be divided into three stages based on reaction progress:
Exponential amplification: At every cycle, the amount of product is doubled (assuming 100% reaction efficiency). After 30 cycles, a single copy of DNA can be increased up to 1,000,000,000 (one billion) copies. In a sense, then, the replication of a discrete strand of DNA is being manipulated in a tube under controlled conditions. The reaction is very sensitive: only minute quantities of DNA must be present.
Leveling off stage: The reaction slows as the DNA polymerase loses activity and as consumption of reagents, such as dNTPs and primers, causes them to become more limited.
Plateau: No more product accumulates due to exhaustion of reagents and enzyme.
In practice, PCR can fail for various reasons, in part due to its sensitivity to contamination causing amplification of spurious DNA products. Because of this, a number of techniques and procedures have been developed for optimizing PCR conditions. Contamination with extraneous DNA is addressed with lab protocols and procedures that separate pre-PCR mixtures from potential DNA contaminants. This usually involves spatial separation of PCR-setup areas from areas for analysis or purification of PCR products, use of disposable plasticware, and thoroughly cleaning the work surface between reaction setups. Primer-design techniques are important in improving PCR product yield and in avoiding the formation of spurious products, and the usage of alternate buffer components or polymerase enzymes can help with amplification of long or otherwise problematic regions of DNA. Addition of reagents, such as formamide, in buffer systems may increase the specificity and yield of PCR. Computer simulations of theoretical PCR results (Electronic PCR) may be performed to assist in primer design
This is a visualization of Exponential Amplification to give a better understanding of what it is and why it is so important in the PCR process. Showing how it makes copies of the sample collected at the scene.
PCR allows isolation of DNA fragments from genomic DNA by selective amplification of a specific region of DNA. This use of PCR augments many ways, such as generating hybridization probes for Southern or northern hybridization and DNA cloning, which require larger amounts of DNA, representing a specific DNA region. PCR supplies these techniques with high amounts of pure DNA, enabling analysis of DNA samples even from very small amounts of starting material.
Other applications of PCR include DNA sequencing to determine unknown PCR-amplified sequences in which one of the amplification primers may be used in Sanger sequencing, isolation of a DNA sequence to expedite recombinant DNA technologies involving the insertion of a DNA sequence into a plasmid, phage, or cosmid (depending on size) or the genetic material of another organism. Bacterial colonies (such as E. coli) can be rapidly screened by PCR for correct DNA vector constructs. PCR may also be used for genetic fingerprinting; a forensic technique used to identify a person or organism by comparing experimental DNAs through different PCR-based methods.
Some PCR 'fingerprints' methods have high discriminative power and can be used to identify genetic relationships between individuals, such as parent-child or between siblings, and are used in paternity testing (Fig. 4). This technique may also be used to determine evolutionary relationships among organisms when certain molecular clocks are used (i.e., the 16S rRNA and recA genes of microorganisms).
Because PCR amplifies the regions of DNA that it targets, PCR can be used to analyze extremely small amounts of sample. This is often critical for forensic analysis, when only a trace amount of DNA is available as evidence. PCR may also be used in the analysis of ancient DNA that is tens of thousands of years old. These PCR-based techniques have been successfully used on animals, such as a forty-thousand-year-old mammoth, and also on human DNA, in applications ranging from the analysis of Egyptian mummies to the identification of a Russiantsar and the body of English king Richard III.
Quantitative PCR or Real Time PCR (qPCR, not to be confused with RT-PCR) methods allow the estimation of the amount of a given sequence present in a sample—a technique often applied to quantitatively determine levels of gene expression. Quantitative PCR is an established tool for DNA quantification that measures the accumulation of DNA product after each round of PCR amplification.
qPCR allows the quantification and detection of a specific DNA sequence in real time since it measures concentration while the synthesis process is taking place. There are two methods for simultaneous detection and quantification. The first method consists of using fluorescent dyes that are retained nonspecifically in between the double strands. The second method involves probes that code for specific sequences and are fluorescently labeled. Detection of DNA using these methods can only be seen after the hybridization of probes with its complementary DNA takes place. An interesting technique combination is real-time PCR and reverse transcription. This sophisticated technique, called RT-qPCR, allows for the quantification of a small quantity of RNA. Through this combined technique, mRNA is converted to cDNA, which is further quantified using qPCR. This technique lowers the possibility of error at the end point of PCR, increasing chances for detection of genes associated with genetic diseases such as cancer. Laboratories use RT-qPCR for the purpose of sensitively measuring gene regulation.
Electrophoresis of PCR-amplified DNA fragments. (1) Father. (2) Child. (3) Mother. The child has inherited some, but not all of the fingerprints of each of its parents, giving it a new, unique fingerprint.
Prospective parents can be tested for being genetic carriers, or their children might be tested for actually being affected by a disease. DNA samples for prenatal testing can be obtained by amniocentesis, chorionic villus sampling, or even by the analysis of rare fetal cells circulating in the mother's bloodstream. PCR analysis is also essential to preimplantation genetic diagnosis, where individual cells of a developing embryo are tested for mutations.
PCR can also be used as part of a sensitive test for tissue typing, vital to organ transplantation. As of 2008, there is even a proposal to replace the traditional antibody-based tests for blood type with PCR-based tests.
Many forms of cancer involve alterations to oncogenes. By using PCR-based tests to study these mutations, therapy regimens can sometimes be individually customized to a patient. PCR permits early diagnosis of malignant diseases such as leukemia and lymphomas, which is currently the highest-developed in cancer research and is already being used routinely. PCR assays can be performed directly on genomic DNA samples to detect translocation-specific malignant cells at a sensitivity that is at least 10,000 fold higher than that of other methods. PCR is very useful in the medical field since it allows for the isolation and amplification of tumor suppressors. Quantitative PCR for example, can be used to quantify and analyze single cells, as well as recognize DNA, mRNA and protein confirmations and combinations.
PCR allows for rapid and highly specific diagnosis of infectious diseases, including those caused by bacteria or viruses. PCR also permits identification of non-cultivatable or slow-growing microorganisms such as mycobacteria, anaerobic bacteria, or viruses from tissue culture assays and animal models. The basis for PCR diagnostic applications in microbiology is the detection of infectious agents and the discrimination of non-pathogenic from pathogenic strains by virtue of specific genes.
Characterization and detection of infectious disease organisms have been revolutionized by PCR in the following ways:
The human immunodeficiency virus (or HIV), is a difficult target to find and eradicate. The earliest tests for infection relied on the presence of antibodies to the virus circulating in the bloodstream. However, antibodies don't appear until many weeks after infection, maternal antibodies mask the infection of a newborn, and therapeutic agents to fight the infection don't affect the antibodies. PCR tests have been developed that can detect as little as one viral genome among the DNA of over 50,000 host cells. Infections can be detected earlier, donated blood can be screened directly for the virus, newborns can be immediately tested for infection, and the effects of antiviral treatments can be quantified.
Some disease organisms, such as that for tuberculosis, are difficult to sample from patients and slow to be grown in the laboratory. PCR-based tests have allowed detection of small numbers of disease organisms (both live or dead), in convenient samples. Detailed genetic analysis can also be used to detect antibiotic resistance, allowing immediate and effective therapy. The effects of therapy can also be immediately evaluated.
The spread of a disease organism through populations of domestic or wild animals can be monitored by PCR testing. In many cases, the appearance of new virulent sub-types can be detected and monitored. The sub-types of an organism that were responsible for earlier epidemics can also be determined by PCR analysis.
Viral DNA can be detected by PCR. The primers used must be specific to the targeted sequences in the DNA of a virus, and PCR can be used for diagnostic analyses or DNA sequencing of the viral genome. The high sensitivity of PCR permits virus detection soon after infection and even before the onset of disease. Such early detection may give physicians a significant lead time in treatment. The amount of virus ("viral load") in a patient can also be quantified by PCR-based DNA quantitation techniques (see below). A variant of PCR ( RT-PCR) is used for detecting viral RNA rather than DNA: in this test the enzyme reverse transcriptase is used to generate a DNA sequence which matches the viral RNA; this DNA is then amplified as per the usual PCR method. RT-PCR is widely used to detect the Sars-Cov-2 viral genome.
Diseases such as pertussis (or whooping cough) are caused by the bacteria Bordetella pertussis. This bacteria is marked by a serious acute respiratory infection that affects various animals and humans and has led to the deaths of many young children. The pertussis toxin is a protein exotoxin that binds to cell receptors by two dimers and reacts with different cell types such as T lymphocytes which play a role in cell immunity. PCR is an important testing tool that can detect sequences within the gene for the pertussis toxin. Because PCR has a high sensitivity for the toxin and a rapid turnaround time, it is very efficient for diagnosing pertussis when compared to culture.
The development of PCR-based genetic (or DNA) fingerprinting protocols has seen widespread application in forensics:
In its most discriminating form, genetic fingerprinting can uniquely discriminate any one person from the entire population of the world. Minute samples of DNA can be isolated from a crime scene, and compared to that from suspects, or from a DNA database of earlier evidence or convicts. Simpler versions of these tests are often used to rapidly rule out suspects during a criminal investigation. Evidence from decades-old crimes can be tested, confirming or exonerating the people originally convicted.
Forensic DNA typing has been an effective way of identifying or exonerating criminal suspects due to analysis of evidence discovered at a crime scene. The human genome has many repetitive regions that can be found within gene sequences or in non-coding regions of the genome. Specifically, up to 40% of human DNA is repetitive. There are two distinct categories for these repetitive, non-coding regions in the genome. The first category is called variable number tandem repeats (VNTR), which are 10–100 base pairs long and the second category is called short tandem repeats (STR) and these consist of repeated 2–10 base pair sections. PCR is used to amplify several well-known VNTRs and STRs using primers that flank each of the repetitive regions. The sizes of the fragments obtained from any individual for each of the STRs will indicate which alleles are present. By analyzing several STRs for an individual, a set of alleles for each person will be found that statistically is likely to be unique. Researchers have identified the complete sequence of the human genome. This sequence can be easily accessed through the NCBI website and is used in many real-life applications. For example, the FBI has compiled a set of DNA marker sites used for identification, and these are called the Combined DNA Index System (CODIS) DNA database. Using this database enables statistical analysis to be used to determine the probability that a DNA sample will match. PCR is a very powerful and significant analytical tool to use for forensic DNA typing because researchers only need a very small amount of the target DNA to be used for analysis. For example, a single human hair with attached hair follicle has enough DNA to conduct the analysis. Similarly, a few sperm, skin samples from under the fingernails, or a small amount of blood can provide enough DNA for conclusive analysis.
Less discriminating forms of DNA fingerprinting can help in DNA paternity testing, where an individual is matched with their close relatives. DNA from unidentified human remains can be tested, and compared with that from possible parents, siblings, or children. Similar testing can be used to confirm the biological parents of an adopted (or kidnapped) child. The actual biological father of a newborn can also be confirmed (or ruled out).
The PCR AMGX/AMGY design has been shown to not only facilitate in amplifying DNA sequences from a very minuscule amount of genome. However it can also be used for real time sex determination from forensic bone samples. This provides us with a powerful and effective way to determine the sex of not only ancient specimens but also current suspects in crimes.
PCR has been applied to many areas of research in molecular genetics:
PCR allows rapid production of short pieces of DNA, even when not more than the sequence of the two primers is known. This ability of PCR augments many methods, such as generating hybridizationprobes for Southern or northern blot hybridization. PCR supplies these techniques with large amounts of pure DNA, sometimes as a single strand, enabling analysis even from very small amounts of starting material.
The task of DNA sequencing can also be assisted by PCR. Known segments of DNA can easily be produced from a patient with a genetic disease mutation. Modifications to the amplification technique can extract segments from a completely unknown genome, or can generate just a single strand of an area of interest.
PCR has numerous applications to the more traditional process of DNA cloning. It can extract segments for insertion into a vector from a larger genome, which may be only available in small quantities. Using a single set of 'vector primers', it can also analyze or extract fragments that have already been inserted into vectors. Some alterations to the PCR protocol can generate mutations (general or site-directed) of an inserted fragment.
Sequence-tagged sites is a process where PCR is used as an indicator that a particular segment of a genome is present in a particular clone. The Human Genome Project found this application vital to mapping the cosmid clones they were sequencing, and to coordinating the results from different laboratories.
An application of PCR is the phylogenic analysis of DNA from ancient sources, such as that found in the recovered bones of Neanderthals, from frozen tissues of mammoths, or from the brain of Egyptian mummies. In some cases the highly degraded DNA from these sources might be reassembled during the early stages of amplification.
A common application of PCR is the study of patterns of gene expression. Tissues (or even individual cells) can be analyzed at different stages to see which genes have become active, or which have been switched off. This application can also use quantitative PCR to quantitate the actual levels of expression
The ability of PCR to simultaneously amplify several loci from individual sperm has greatly enhanced the more traditional task of genetic mapping by studying chromosomal crossovers after meiosis. Rare crossover events between very close loci have been directly observed by analyzing thousands of individual sperms. Similarly, unusual deletions, insertions, translocations, or inversions can be analyzed, all without having to wait (or pay) for the long and laborious processes of fertilization, embryogenesis, etc.
Site-directed mutagenesis: PCR can be used to create mutant genes with mutations chosen by scientists at will. These mutations can be chosen in order to understand how proteins accomplish their functions, and to change or improve protein function.
PCR has a number of advantages. It is fairly simple to understand and to use, and produces results rapidly. The technique is highly sensitive with the potential to produce millions to billions of copies of a specific product for sequencing, cloning, and analysis. qRT-PCR shares the same advantages as the PCR, with an added advantage of quantification of the synthesized product. Therefore, it has its uses to analyze alterations of gene expression levels in tumors, microbes, or other disease states.
PCR is a very powerful and practical research tool. The sequencing of unknown etiologies of many diseases are being figured out by the PCR. The technique can help identify the sequence of previously unknown viruses related to those already known and thus give us a better understanding of the disease itself. If the procedure can be further simplified and sensitive non radiometric detection systems can be developed, the PCR will assume a prominent place in the clinical laboratory for years to come.
One major limitation of PCR is that prior information about the target sequence is necessary in order to generate the primers that will allow its selective amplification. This means that, typically, PCR users must know the precise sequence(s) upstream of the target region on each of the two single-stranded templates in order to ensure that the DNA polymerase properly binds to the primer-template hybrids and subsequently generates the entire target region during DNA synthesis.
Like all enzymes, DNA polymerases are also prone to error, which in turn causes mutations in the PCR fragments that are generated.
Another limitation of PCR is that even the smallest amount of contaminating DNA can be amplified, resulting in misleading or ambiguous results. To minimize the chance of contamination, investigators should reserve separate rooms for reagent preparation, the PCR, and analysis of product. Reagents should be dispensed into single-use aliquots. Pipettors with disposable plungers and extra-long pipette tips should be routinely used.
Environmental samples that contain humic acids may inhibit PCR amplification and lead to inaccurate results.
Allele-specific PCR: a diagnostic or cloning technique based on single-nucleotide variations (SNVs not to be confused with SNPs) (single-base differences in a patient). It requires prior knowledge of a DNA sequence, including differences between alleles, and uses primers whose 3' ends encompass the SNV (base pair buffer around SNV usually incorporated). PCR amplification under stringent conditions is much less efficient in the presence of a mismatch between template and primer, so successful amplification with an SNP-specific primer signals presence of the specific SNP in a sequence. See SNP genotyping for more information.
Assembly PCR or Polymerase Cycling Assembly (PCA): artificial synthesis of long DNA sequences by performing PCR on a pool of long oligonucleotides with short overlapping segments. The oligonucleotides alternate between sense and antisense directions, and the overlapping segments determine the order of the PCR fragments, thereby selectively producing the final long DNA product.
Asymmetric PCR: preferentially amplifies one DNA strand in a double-stranded DNA template. It is used in sequencing and hybridization probing where amplification of only one of the two complementary strands is required. PCR is carried out as usual, but with a great excess of the primer for the strand targeted for amplification. Because of the slow (arithmetic) amplification later in the reaction after the limiting primer has been used up, extra cycles of PCR are required. A recent modification on this process, known as Linear-After-The-Exponential-PCR (LATE-PCR), uses a limiting primer with a higher melting temperature (Tm) than the excess primer to maintain reaction efficiency as the limiting primer concentration decreases mid-reaction.
Convective PCR: a pseudo-isothermal way of performing PCR. Instead of repeatedly heating and cooling the PCR mixture, the solution is subjected to a thermal gradient. The resulting thermal instability driven convective flow automatically shuffles the PCR reagents from the hot and cold regions repeatedly enabling PCR. Parameters such as thermal boundary conditions and geometry of the PCR enclosure can be optimized to yield robust and rapid PCR by harnessing the emergence of chaotic flow fields. Such convective flow PCR setup significantly reduces device power requirement and operation time.
Dial-out PCR: a highly parallel method for retrieving accurate DNA molecules for gene synthesis. A complex library of DNA molecules is modified with unique flanking tags before massively parallel sequencing. Tag-directed primers then enable the retrieval of molecules with desired sequences by PCR.
Digital PCR (dPCR): used to measure the quantity of a target DNA sequence in a DNA sample. The DNA sample is highly diluted so that after running many PCRs in parallel, some of them do not receive a single molecule of the target DNA. The target DNA concentration is calculated using the proportion of negative outcomes. Hence the name 'digital PCR'.
Helicase-dependent amplification: similar to traditional PCR, but uses a constant temperature rather than cycling through denaturation and annealing/extension cycles. DNA helicase, an enzyme that unwinds DNA, is used in place of thermal denaturation.
Hot start PCR: a technique that reduces non-specific amplification during the initial set up stages of the PCR. It may be performed manually by heating the reaction components to the denaturation temperature (e.g., 95 °C) before adding the polymerase. Specialized enzyme systems have been developed that inhibit the polymerase's activity at ambient temperature, either by the binding of an antibody or by the presence of covalently bound inhibitors that dissociate only after a high-temperature activation step. Hot-start/cold-finish PCR is achieved with new hybrid polymerases that are inactive at ambient temperature and are instantly activated at elongation temperature.
In silico PCR (digital PCR, virtual PCR, electronic PCR, e-PCR) refers to computational tools used to calculate theoretical polymerase chain reaction results using a given set of primers (probes) to amplify DNA sequences from a sequenced genome or transcriptome. In silico PCR was proposed as an educational tool for molecular biology.
Intersequence-specific PCR (ISSR): a PCR method for DNA fingerprinting that amplifies regions between simple sequence repeats to produce a unique fingerprint of amplified fragment lengths.
Inverse PCR: is commonly used to identify the flanking sequences around genomic inserts. It involves a series of DNA digestions and self ligation, resulting in known sequences at either end of the unknown sequence.
Ligation-mediated PCR: uses small DNA linkers ligated to the DNA of interest and multiple primers annealing to the DNA linkers; it has been used for DNA sequencing, genome walking, and DNA footprinting.
Methylation-specific PCR (MSP): developed by Stephen Baylin and James G. Herman at the Johns Hopkins School of Medicine, and is used to detect methylation of CpG islands in genomic DNA. DNA is first treated with sodium bisulfite, which converts unmethylated cytosine bases to uracil, which is recognized by PCR primers as thymine. Two PCRs are then carried out on the modified DNA, using primer sets identical except at any CpG islands within the primer sequences. At these points, one primer set recognizes DNA with cytosines to amplify methylated DNA, and one set recognizes DNA with uracil or thymine to amplify unmethylated DNA. MSP using qPCR can also be performed to obtain quantitative rather than qualitative information about methylation.
Miniprimer PCR: uses a thermostable polymerase (S-Tbr) that can extend from short primers ("smalligos") as short as 9 or 10 nucleotides. This method permits PCR targeting to smaller primer binding regions, and is used to amplify conserved DNA sequences, such as the 16S (or eukaryotic 18S) rRNA gene.
Multiplex-PCR: consists of multiple primer sets within a single PCR mixture to produce amplicons of varying sizes that are specific to different DNA sequences. By targeting multiple genes at once, additional information may be gained from a single test-run that otherwise would require several times the reagents and more time to perform. Annealing temperatures for each of the primer sets must be optimized to work correctly within a single reaction, and amplicon sizes. That is, their base pair length should be different enough to form distinct bands when visualized by gel electrophoresis.
Nanoparticle-Assisted PCR (nanoPCR): some nanoparticles (NPs) can enhance the efficiency of PCR (thus being called nanoPCR), and some can even outperform the original PCR enhancers. It was reported that quantum dots (QDs) can improve PCR specificity and efficiency. Single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) are efficient in enhancing the amplification of long PCR. Carbon nanopowder (CNP) can improve the efficiency of repeated PCR and long PCR, while zinc oxide, titanium dioxide and Ag NPs were found to increase the PCR yield. Previous data indicated that non-metallic NPs retained acceptable amplification fidelity. Given that many NPs are capable of enhancing PCR efficiency, it is clear that there is likely to be great potential for nanoPCR technology improvements and product development.
Nested PCR: increases the specificity of DNA amplification, by reducing background due to non-specific amplification of DNA. Two sets of primers are used in two successive PCRs. In the first reaction, one pair of primers is used to generate DNA products, which besides the intended target, may still consist of non-specifically amplified DNA fragments. The product(s) are then used in a second PCR with a set of primers whose binding sites are completely or partially different from and located 3' of each of the primers used in the first reaction. Nested PCR is often more successful in specifically amplifying long DNA fragments than conventional PCR, but it requires more detailed knowledge of the target sequences.
Overlap-extension PCR or Splicing by overlap extension (SOEing) : a genetic engineering technique that is used to splice together two or more DNA fragments that contain complementary sequences. It is used to join DNA pieces containing genes, regulatory sequences, or mutations; the technique enables creation of specific and long DNA constructs. It can also introduce deletions, insertions or point mutations into a DNA sequence.
PAN-AC: uses isothermal conditions for amplification, and may be used in living cells.
quantitative PCR (qPCR): used to measure the quantity of a target sequence (commonly in real-time). It quantitatively measures starting amounts of DNA, cDNA, or RNA. quantitative PCR is commonly used to determine whether a DNA sequence is present in a sample and the number of its copies in the sample. Quantitative PCR has a very high degree of precision. Quantitative PCR methods use fluorescent dyes, such as Sybr Green, EvaGreen or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product in real time. It is also sometimes abbreviated to RT-PCR (real-time PCR) but this abbreviation should be used only for reverse transcription PCR. qPCR is the appropriate contractions for quantitative PCR (real-time PCR).
Reverse Transcription PCR (RT-PCR): for amplifying DNA from RNA. Reverse transcriptase reverse transcribes RNA into cDNA, which is then amplified by PCR. RT-PCR is widely used in expression profiling, to determine the expression of a gene or to identify the sequence of an RNA transcript, including transcription start and termination sites. If the genomic DNA sequence of a gene is known, RT-PCR can be used to map the location of exons and introns in the gene. The 5' end of a gene (corresponding to the transcription start site) is typically identified by RACE-PCR (Rapid Amplification of cDNA Ends).
RNase H-dependent PCR (rhPCR): a modification of PCR that utilizes primers with a 3’ extension block that can be removed by a thermostable RNase HII enzyme. This system reduces primer-dimers and allows for multiplexed reactions to be performed with higher numbers of primers.
Single Specific Primer-PCR (SSP-PCR): allows the amplification of double-stranded DNA even when the sequence information is available at one end only. This method permits amplification of genes for which only a partial sequence information is available, and allows unidirectional genome walking from known into unknown regions of the chromosome.
Solid Phase PCR: encompasses multiple meanings, including Polony Amplification (where PCR colonies are derived in a gel matrix, for example), Bridge PCR (primers are covalently linked to a solid-support surface), conventional Solid Phase PCR (where Asymmetric PCR is applied in the presence of solid support bearing primer with sequence matching one of the aqueous primers) and Enhanced Solid Phase PCR (where conventional Solid Phase PCR can be improved by employing high Tm and nested solid support primer with optional application of a thermal 'step' to favour solid support priming).
Suicide PCR: typically used in paleogenetics or other studies where avoiding false positives and ensuring the specificity of the amplified fragment is the highest priority. It was originally described in a study to verify the presence of the microbe Yersinia pestis in dental samples obtained from 14th Century graves of people supposedly killed by the plague during the medieval Black Death epidemic. The method prescribes the use of any primer combination only once in a PCR (hence the term "suicide"), which should never have been used in any positive control PCR reaction, and the primers should always target a genomic region never amplified before in the lab using this or any other set of primers. This ensures that no contaminating DNA from previous PCR reactions is present in the lab, which could otherwise generate false positives.
Thermal asymmetric interlaced PCR (TAIL-PCR): for isolation of an unknown sequence flanking a known sequence. Within the known sequence, TAIL-PCR uses a nested pair of primers with differing annealing temperatures; a degenerate primer is used to amplify in the other direction from the unknown sequence.
Touchdown PCR (Step-down PCR): a variant of PCR that aims to reduce nonspecific background by gradually lowering the annealing temperature as PCR cycling progresses. The annealing temperature at the initial cycles is usually a few degrees (3–5 °C) above the Tm of the primers used, while at the later cycles, it is a few degrees (3–5 °C) below the primer Tm. The higher temperatures give greater specificity for primer binding, and the lower temperatures permit more efficient amplification from the specific products formed during the initial cycles.
Universal Fast Walking: for genome walking and genetic fingerprinting using a more specific 'two-sided' PCR than conventional 'one-sided' approaches (using only one gene-specific primer and one general primer—which can lead to artefactual 'noise') by virtue of a mechanism involving lariat structure formation. Streamlined derivatives of UFW are LaNe RAGE (lariat-dependent nested PCR for rapid amplification of genomic DNA ends), 5'RACE LaNe and 3'RACE LaNe.
Diagrammatic representation of an example primer pair. The use of primers in an in vitro assay to allow DNA synthesis was a major innovation that allowed the development of PCR.
Diagrammatic representation of the primers for PCR, indicating the forward and reverse primers and the reverse complement sequence of the reverse primer. (W)
The heat-resistant enzymes that are a key component in polymerase chain reaction were discovered in the 1960s as a product of a microbial life form that lived in the superheated waters of Yellowstone’s Mushroom Spring.
A 1971 paper in the Journal of Molecular Biology by Kjell Kleppe and co-workers in the laboratory of H. Gobind Khorana first described a method of using an enzymatic assay to replicate a short DNA template with primers in vitro. However, this early manifestation of the basic PCR principle did not receive much attention at the time and the invention of the polymerase chain reaction in 1983 is generally credited to Kary Mullis.
When Mullis developed the PCR in 1983, he was working in Emeryville, California for Cetus Corporation, one of the first biotechnology companies, where he was responsible for synthesizing short chains of DNA. Mullis has written that he conceived the idea for PCR while cruising along the Pacific Coast Highway one night in his car. He was playing in his mind with a new way of analyzing changes (mutations) in DNA when he realized that he had instead invented a method of amplifying any DNA region through repeated cycles of duplication driven by DNA polymerase. In Scientific American, Mullis summarized the procedure: "Beginning with a single molecule of the genetic material DNA, the PCR can generate 100 billion similar molecules in an afternoon. The reaction is easy to execute. It requires no more than a test tube, a few simple reagents, and a source of heat." DNA fingerprinting was first used for paternity testing in 1988.
Mullis was awarded the Nobel Prize in Chemistry in 1993 for his invention, seven years after he and his colleagues at Cetus first put his proposal to practice. Mullis's 1985 paper with R. K. Saiki and H. A. Erlich, “Enzymatic Amplification of β-globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia”—the polymerase chain reaction invention (PCR) – was honored by a Citation for Chemical Breakthrough Award from the Division of History of Chemistry of the American Chemical Society in 2017.
At the core of the PCR method is the use of a suitable DNA polymerase able to withstand the high temperatures of >90 °C (194 °F) required for separation of the two DNA strands in the DNA double helix after each replication cycle. The DNA polymerases initially employed for in vitro experiments presaging PCR were unable to withstand these high temperatures. So the early procedures for DNA replication were very inefficient and time-consuming, and required large amounts of DNA polymerase and continuous handling throughout the process.
The discovery in 1976 of Taq polymerase—a DNA polymerase purified from the thermophilic bacterium, Thermus aquaticus, which naturally lives in hot (50 to 80 °C (122 to 176 °F)) environments such as hot springs—paved the way for dramatic improvements of the PCR method. The DNA polymerase isolated from T. aquaticus is stable at high temperatures remaining active even after DNA denaturation, thus obviating the need to add new DNA polymerase after each cycle. This allowed an automated thermocycler-based process for DNA amplification.
"Baby Blue", a 1986 prototype machine for doing PCR.
The PCR technique was patented by Kary Mullis and assigned to Cetus Corporation, where Mullis worked when he invented the technique in 1983. The Taq polymerase enzyme was also covered by patents. There have been several high-profile lawsuits related to the technique, including an unsuccessful lawsuit brought by DuPont. The Swiss pharmaceutical company Hoffmann-La Roche purchased the rights to the patents in 1992 and currently holds those that are still protected.
A related patent battle over the Taq polymerase enzyme is still ongoing in several jurisdictions around the world between Roche and Promega. The legal arguments have extended beyond the lives of the original PCR and Taq polymerase patents, which expired on March 28, 2005.
Polymerase chain reaction ( PCR), a technique used to make numerous copies of a specific segment of DNA quickly and accurately. The polymerase chain reaction enables investigators to obtain the large quantities of DNA that are required for various experiments and procedures in molecular biology, forensic analysis, evolutionary biology, and medical diagnostics.
📹 Learn how DNA thermal cycler employs polymerase chain reaction to copy DNA strands (VİDEO)
📹 Learn how DNA thermal cycler employs polymerase chain reaction to copy DNA strands (LINK)
Learn how DNA thermal cycler employs polymerase chain reaction to copy DNA strands
Specific segments of DNA are amplified (copied) in a laboratory using polymerase chain reaction (PCR) techniques
📂 TRANSCRIPT
NARRATOR: The technique known as the polymerase chain reaction—or, more simply, DNA amplification—enables scientists to prepare genetic material from almost any biological forensic sample. This technique has been automated and is now usually done using an instrument known as the DNA thermal cycler.
First, the sample under investigation is treated to dissolve its DNA content. The solution is then heated, which causes the double strands of the DNA molecule to separate. DNA-polymerizing enzyme is added. This uses each single strand of the original DNA as a template to build up two new copies, so there are now two identical double-stranded molecules of DNA.
A second heating cycle separates the two double strands, and the polymerizing enzyme gets to work again. So now there are four double-stranded molecules of DNA.
With each cycle the number of DNA molecules is doubled. Millions of copies of the original DNA molecule can be synthesized in a few hours.
📹 bio DNA Learn how to DNA fingerprint using agarose gel, Southern blotting, and a radioactive DNA probe / Britannica (VİDEO)
📹 Learn how to DNA fingerprint using agarose gel, Southern blotting, and a radioactive DNA probe / Britannica (LINK)
Learn how to DNA fingerprint using agarose gel, Southern blotting, and a radioactive DNA probe
DNA is extracted, treated with restriction enzymes, and sequenced using gel electrophoresis...
📂 TRANSCRIPT
NARRATOR: Obtaining a genetic fingerprint begins with a sample of biological tissue. At Guy's Hospital, in London, this research worker is using an automatic DNA extractor to produce a high-purity DNA fraction from a blood sample.
A restriction enzyme is added to the DNA. This enzyme moves along the DNA strand, cutting it at the so-called restriction sites. The restriction sites available to enzymes are located throughout the chromosome except in sections consisting of copies of the core sequence. This process produces many pieces of DNA, which consist of repeats of the core sequence.
The mixture of these DNA fragments is then placed onto an agarose gel.
A small voltage is applied across the gel, and fragments move through the gel at a rate proportional to their size. Because gels are difficult to handle, the DNA band pattern on the gel is transferred to a nylon membrane by technique known as Southern blotting.
First, the nylon membrane is placed on the agarose gel. Then, absorbent paper is layered on top of the membrane. It acts as a wick, drawing up the DNA bands onto the membrane, where they stick.
Finally, the researcher adds a radioactive DNA probe, which will bind specifically to the core sequence. Excess DNA probe is washed off, leaving only the radioactive probe that has bound to the DNA pattern on the membrane.
This can be shown on X-ray film, producing the genetic fingerprint.
Polymerase chain reaction
The three-step process of the polymerase chain reaction.
PCR was developed in 1983 by Kary B. Mullis, an American biochemist who won the Nobel Prize for Chemistry in 1993 for his invention. Before the development of PCR, the methods used to amplify, or generate copies of, recombinant DNA fragments were time-consuming and labour-intensive. In contrast, a machine designed to carry out PCR reactions can complete many rounds of replication, producing billions of copies of a DNA fragment, in only a few hours.
The PCR technique is based on the natural processes a cell uses to replicate a new DNA strand. Only a few biological ingredients are needed for PCR. The integral component is the template DNA—i.e., the DNA that contains the region to be copied, such as a gene. As little as one DNA molecule can serve as a template. The only information needed for this fragment to be replicated is the sequence of two short regions of nucleotides (the subunits of DNA) at either end of the region of interest. These two short template sequences must be known so that two primers—short stretches of nucleotides that correspond to the template sequences—can be synthesized. The primers bind, or anneal, to the template at their complementary sites and serve as the starting point for copying. DNA synthesis at one primer is directed toward the other, resulting in replication of the desired intervening sequence. Also needed are free nucleotides used to build the new DNA strands and a DNA polymerase, an enzyme that does the building by sequentially adding on free nucleotides according to the instructions of the template.
PCR is a three-step process that is carried out in repeated cycles. The initial step is the denaturation, or separation, of the two strands of the DNA molecule. This is accomplished by heating the starting material to temperatures of about 95 °C (203 °F). Each strand is a template on which a new strand is built. In the second step the temperature is reduced to about 55 °C (131 °F) so that the primers can anneal to the template. In the third step the temperature is raised to about 72 °C (162 °F), and the DNA polymerase begins adding nucleotides onto the ends of the annealed primers. At the end of the cycle, which lasts about five minutes, the temperature is raised and the process begins again. The number of copies doubles after each cycle. Usually 25 to 30 cycles produce a sufficient amount of DNA.
In the original PCR procedure, one problem was that the DNA polymerase had to be replenished after every cycle because it is not stable at the high temperatures needed for denaturation. This problem was solved in 1987 with the discovery of a heat-stable DNA polymerase called Taq, an enzyme isolated from the thermophilic bacteriumThermus aquaticus, which inhabits hot springs. Taq polymerase also led to the invention of the PCR machine.
Because DNA from a wide range of sources can be amplified, the technique has been applied to many fields. PCR is used to diagnose genetic disease and to detect low levels of viral infection. In forensic medicine it is used to analyze minute traces of blood and other tissues in order to identify the donor by his genetic “fingerprint.” The technique has also been used to amplify DNA fragments found in preserved tissues, such as those of a 40,000-year-old frozen woolly mammoth or of a 7,500-year-old human found in a peat bog.



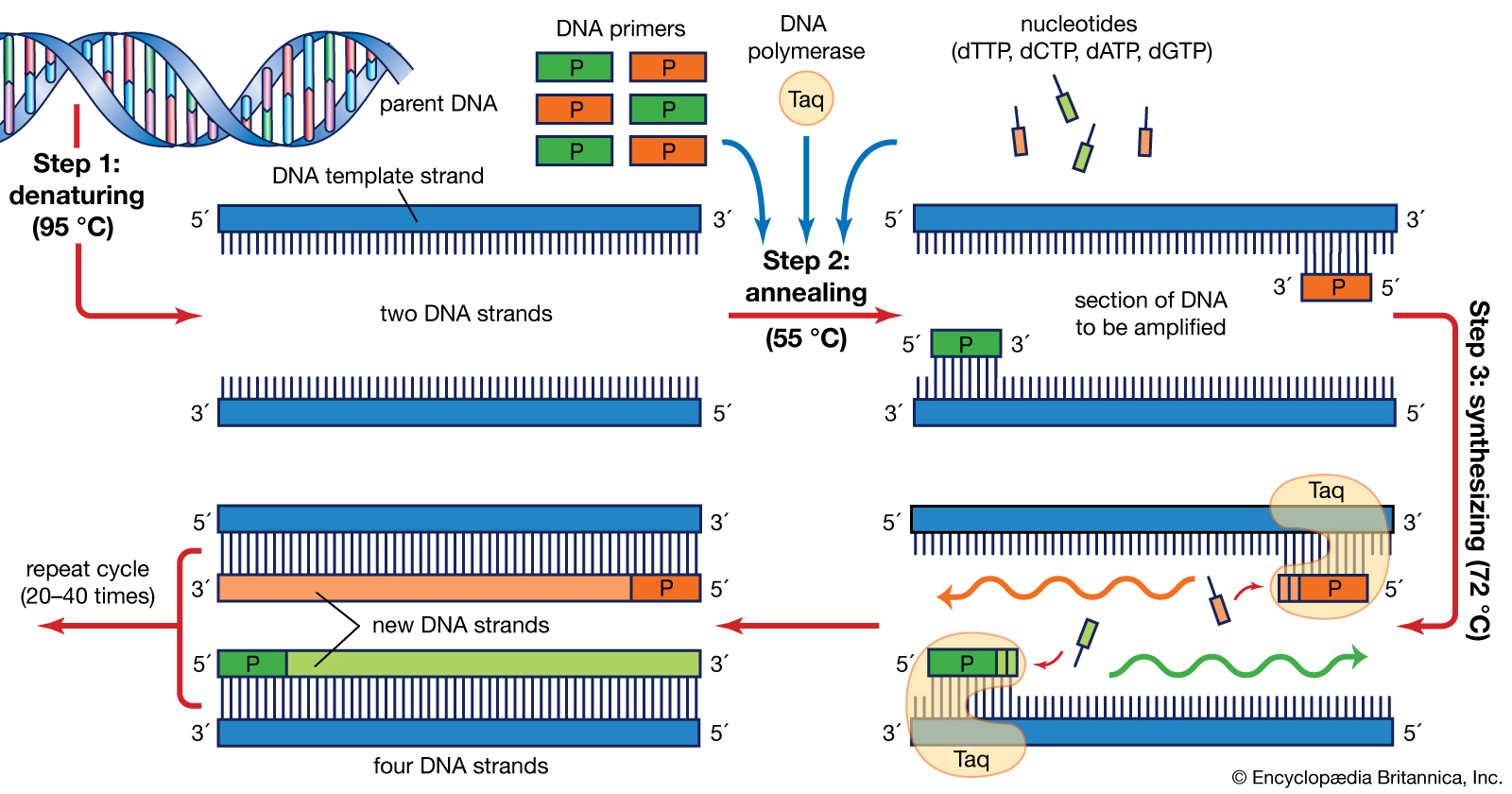
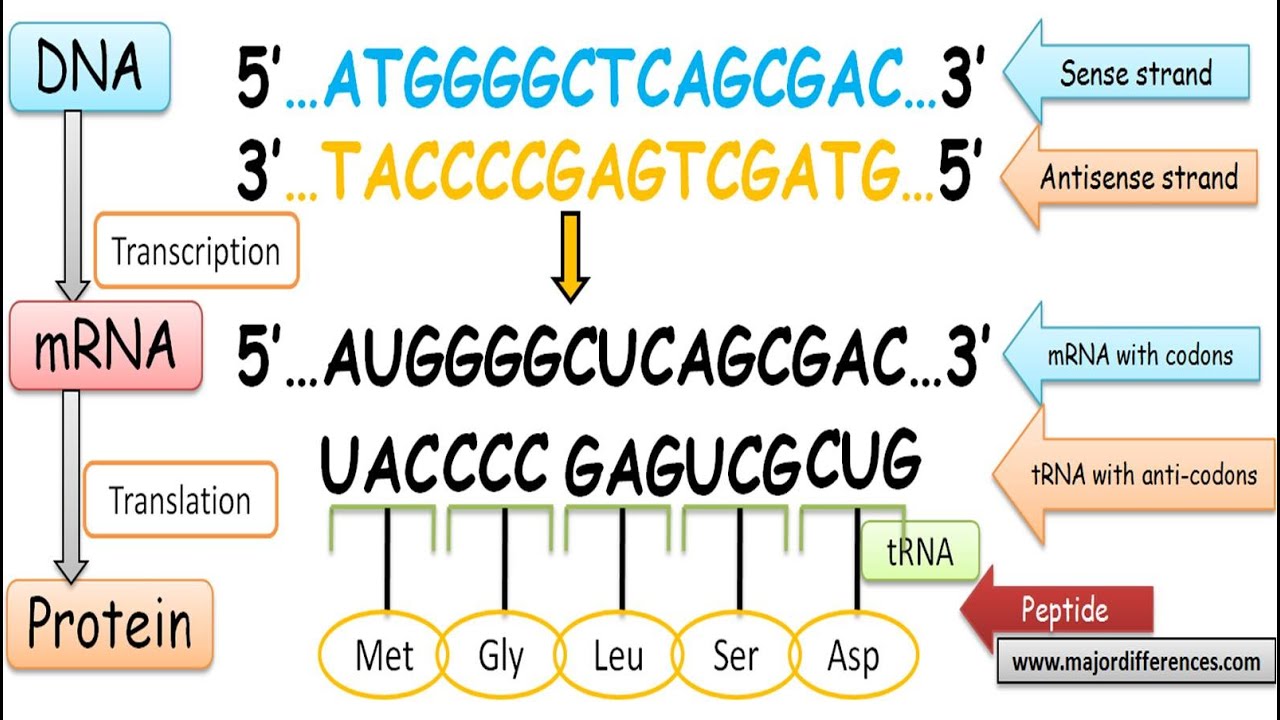
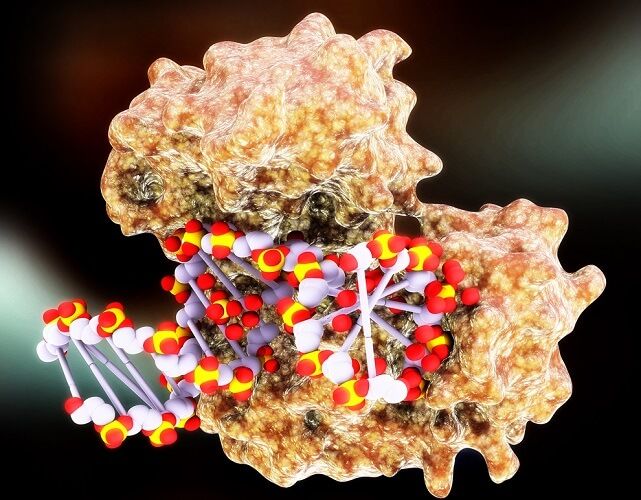
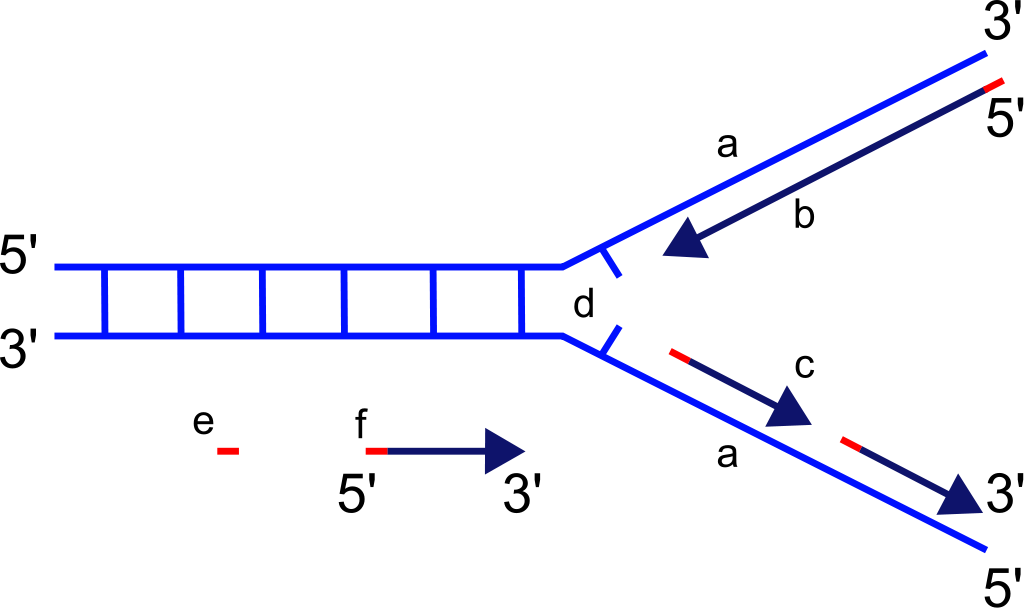
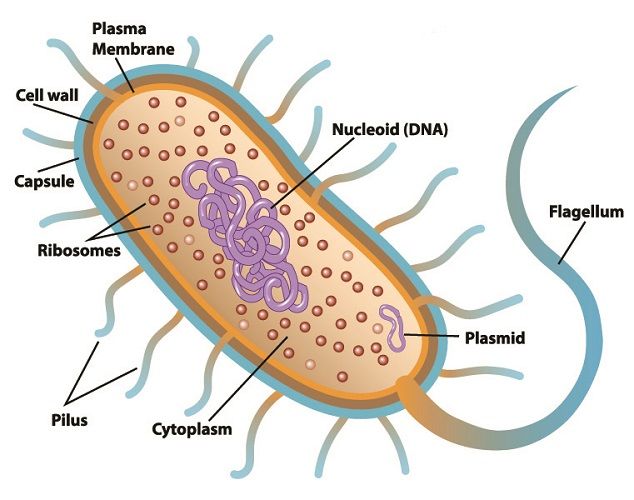
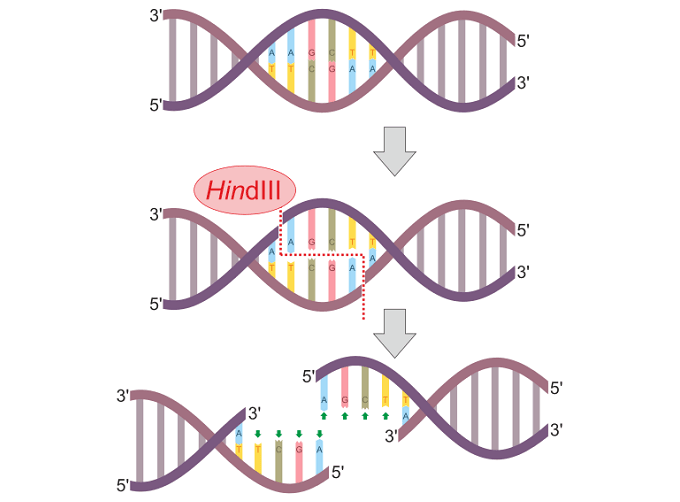
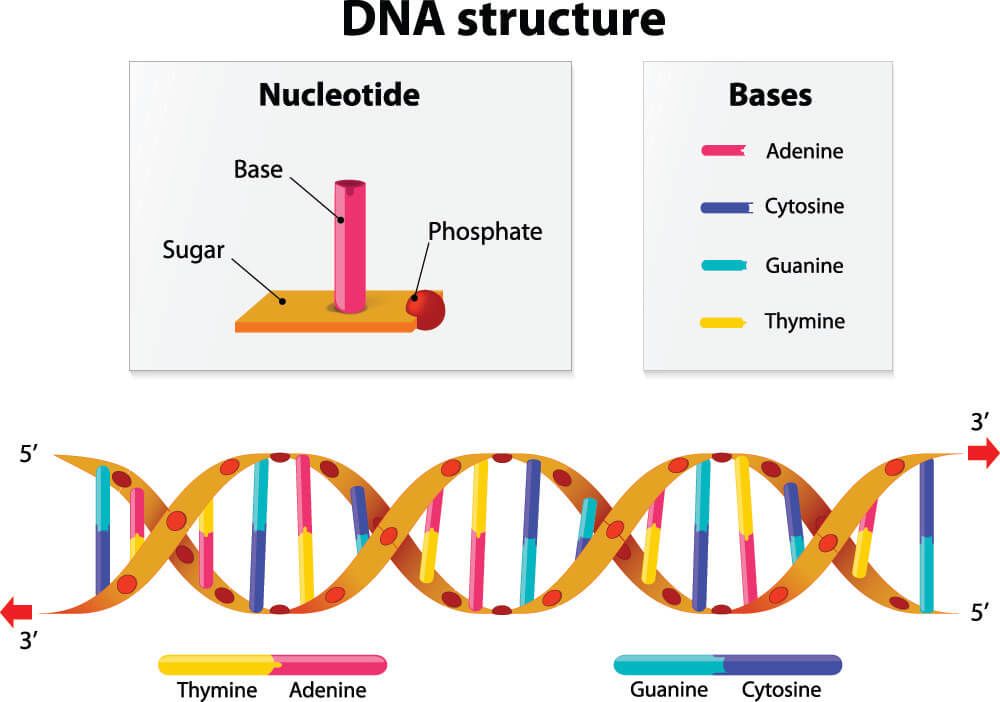
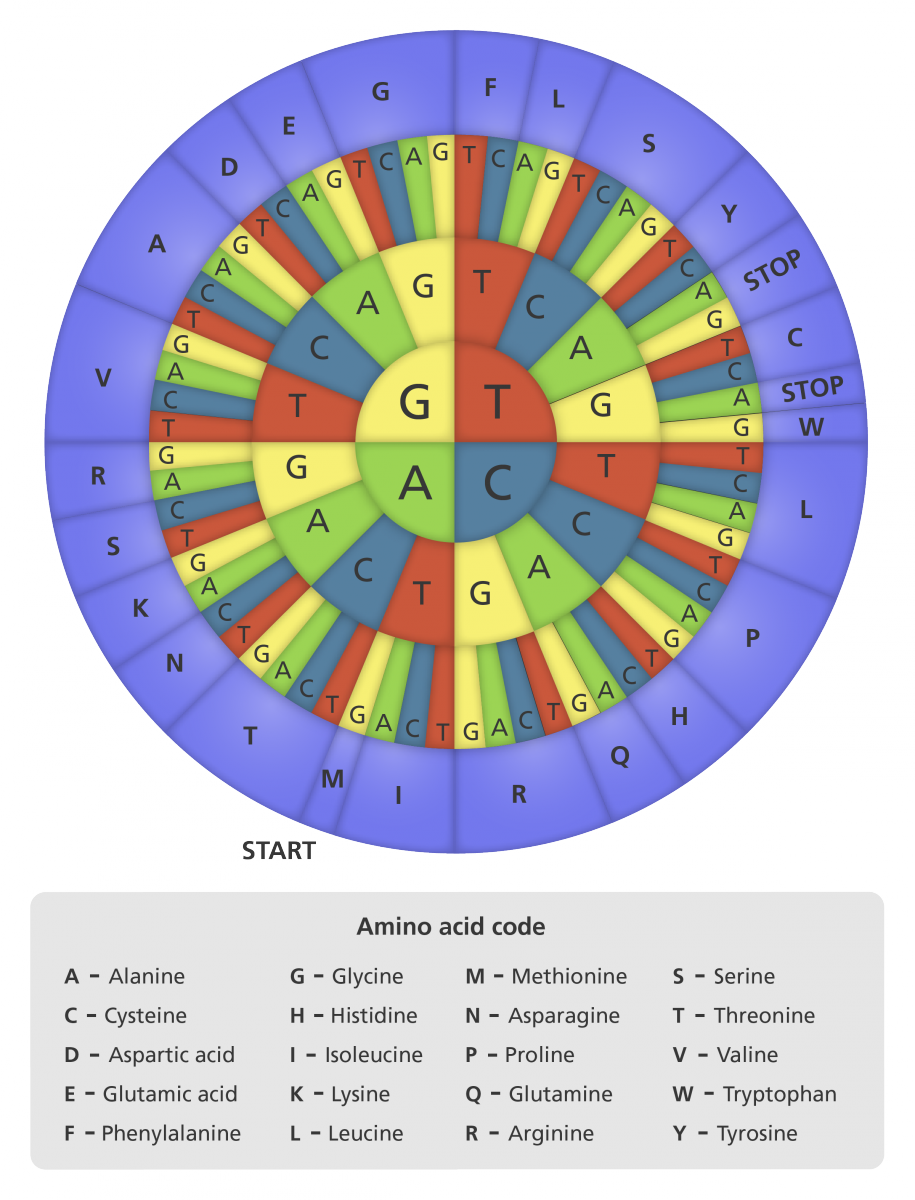
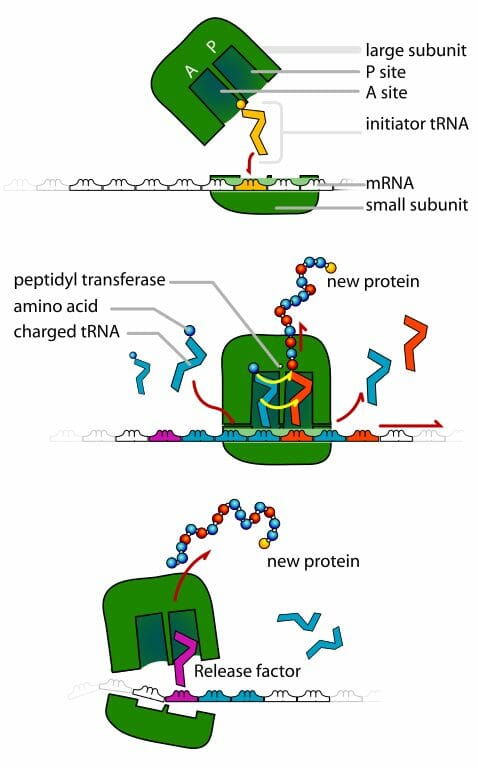
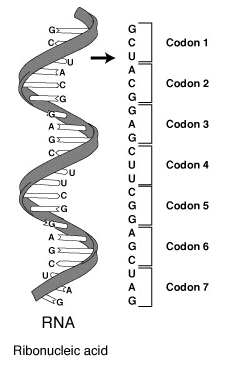
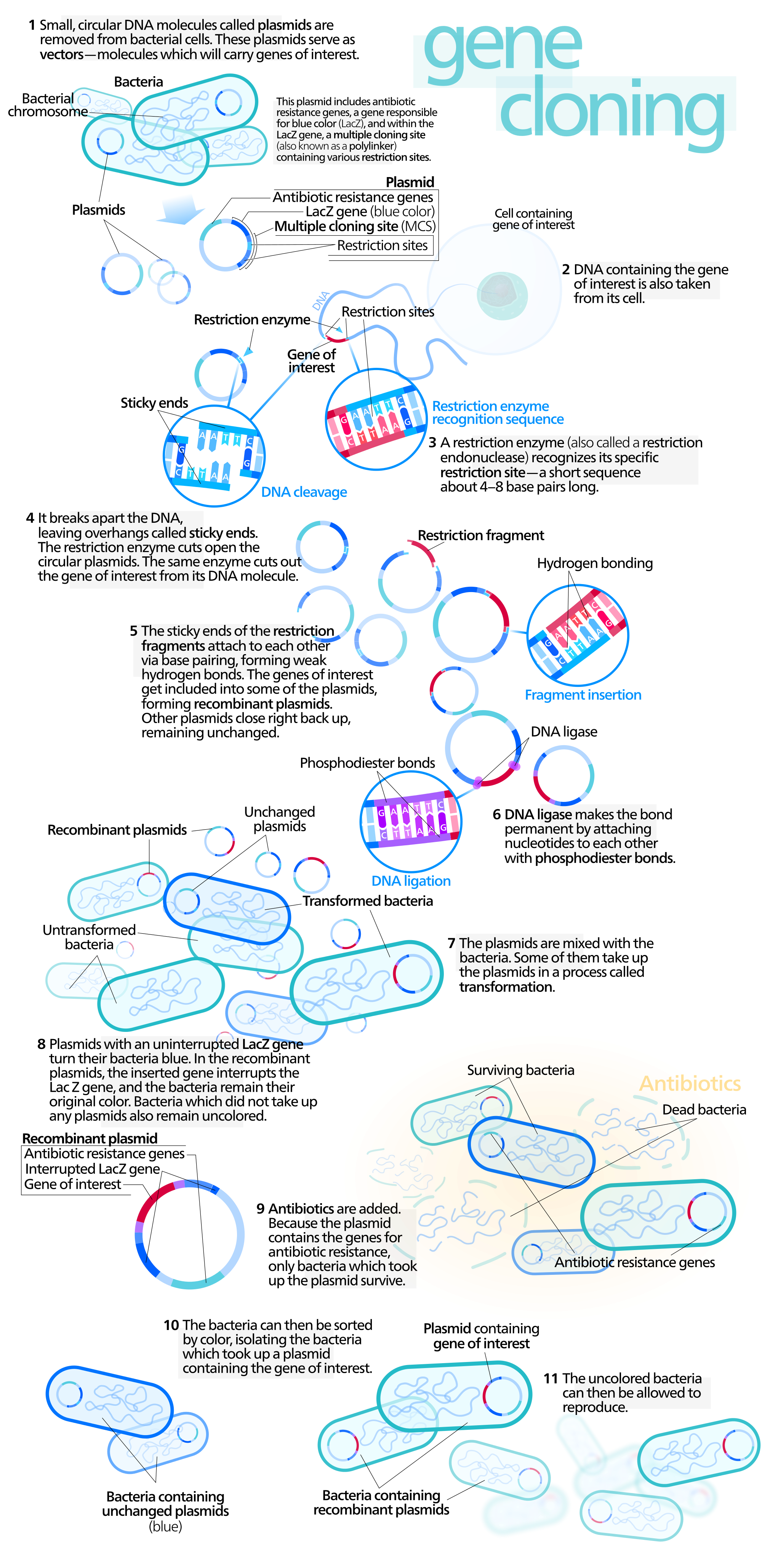
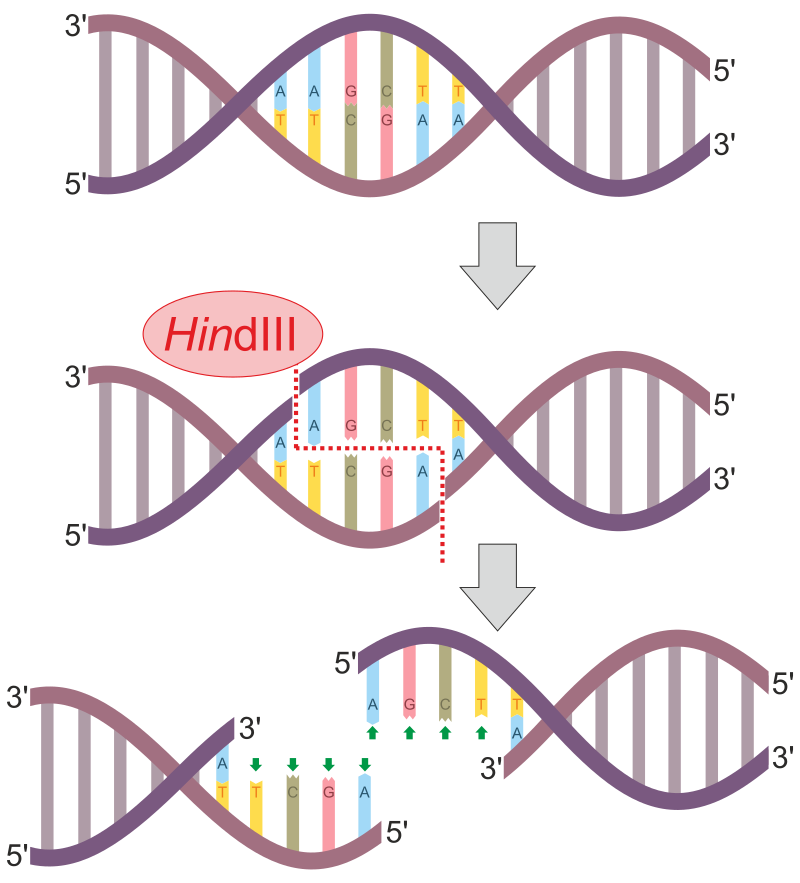
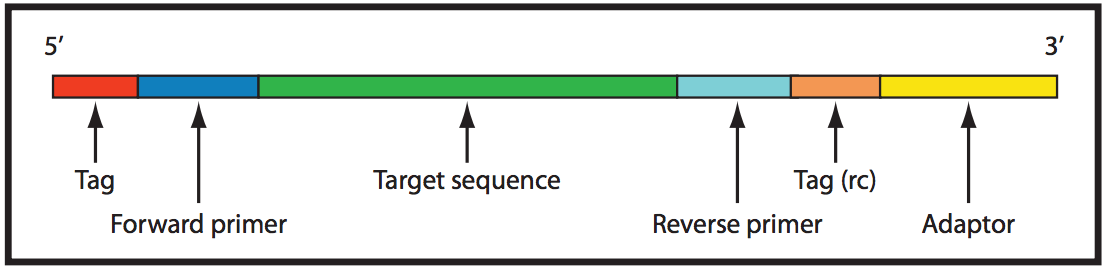
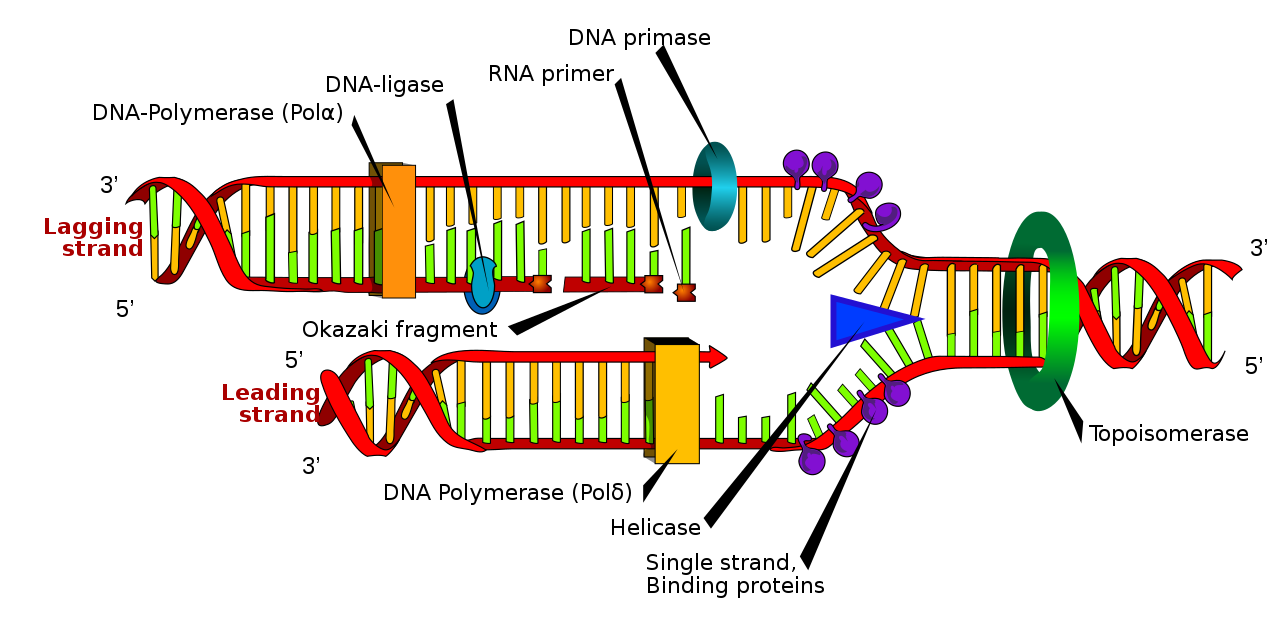
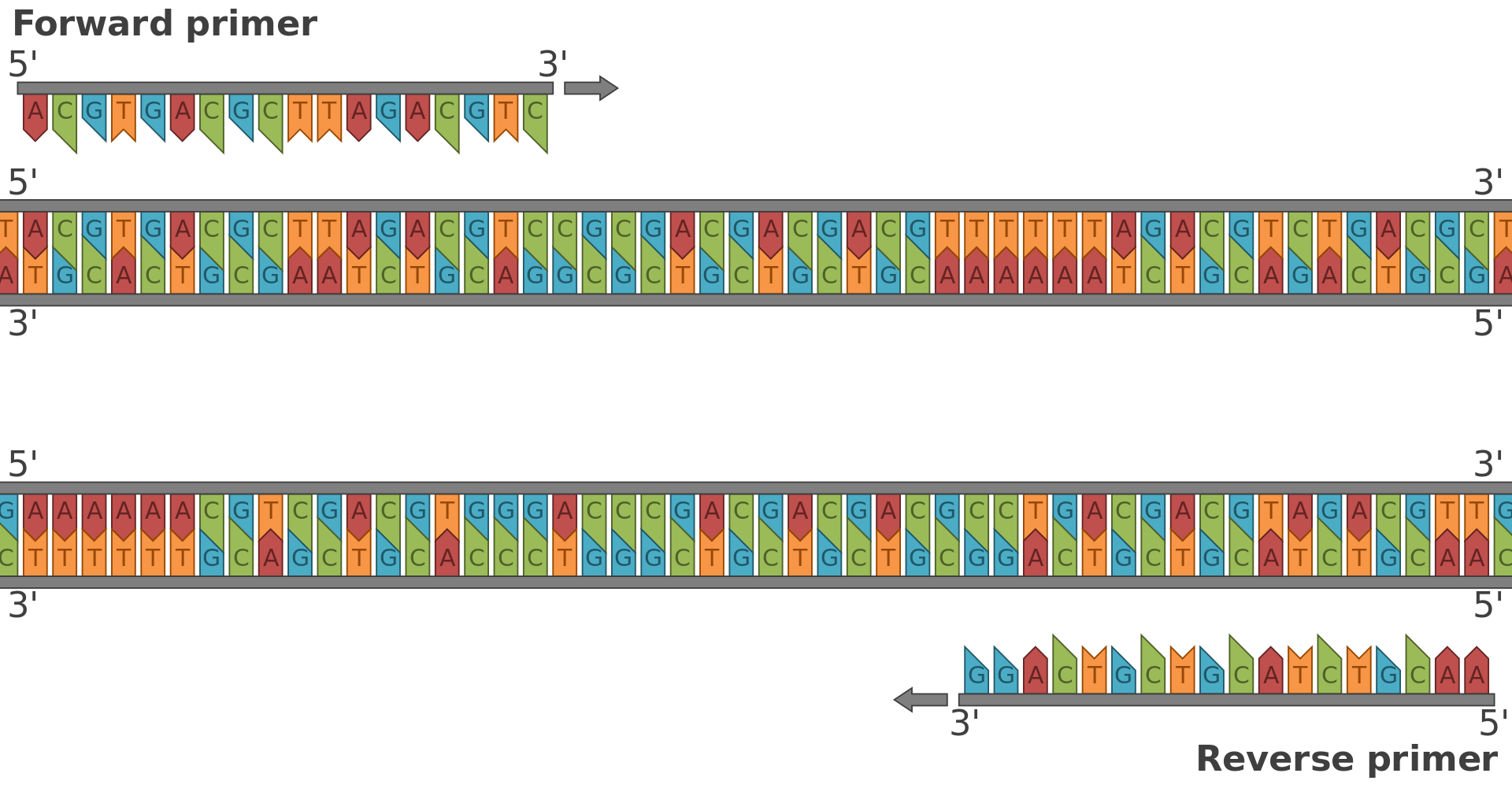
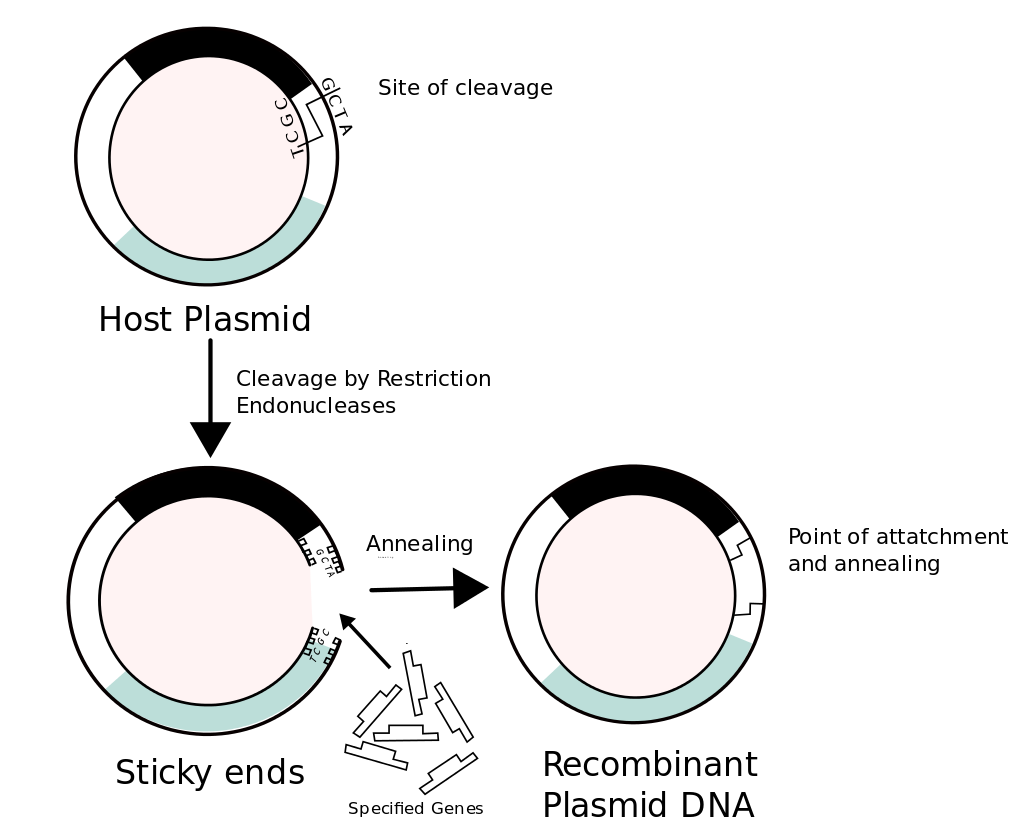
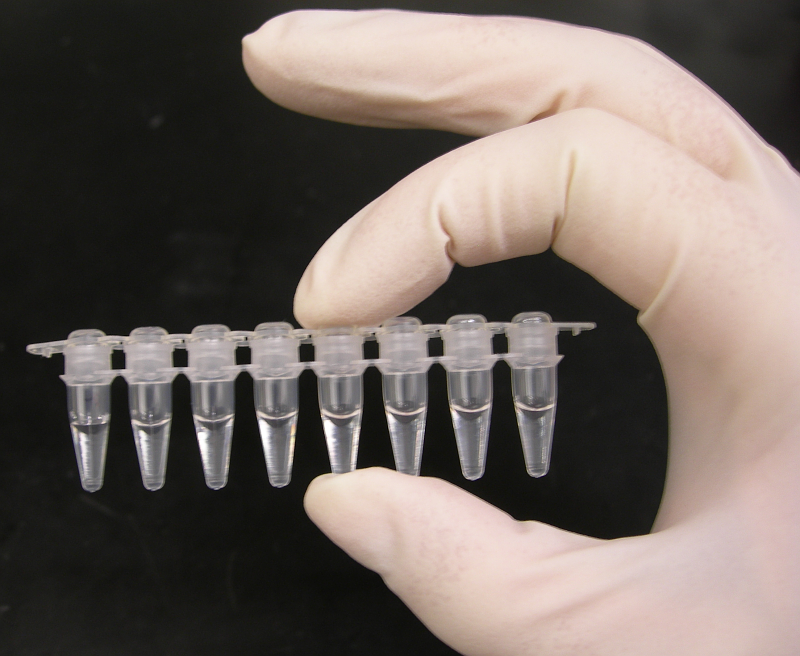

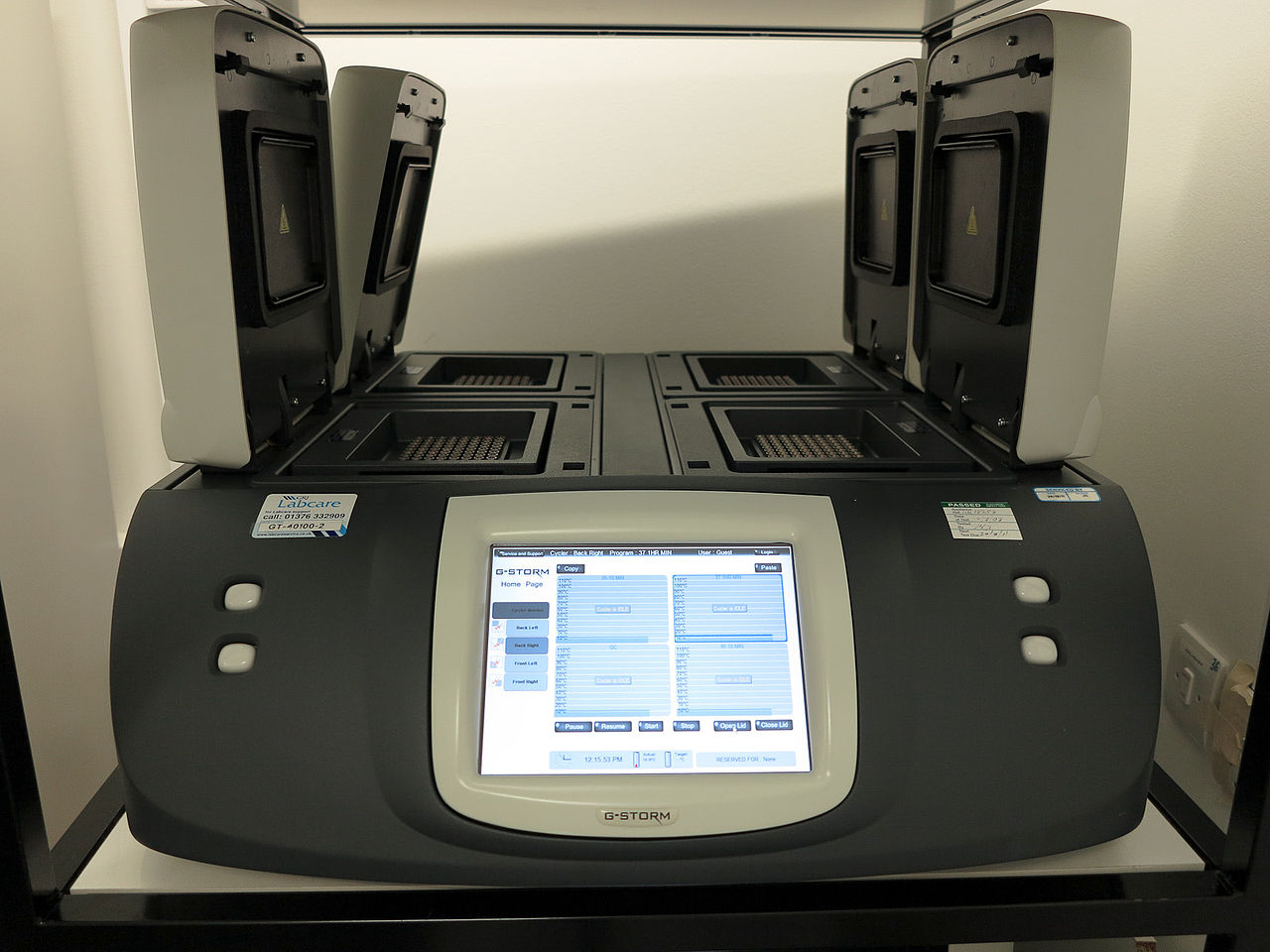

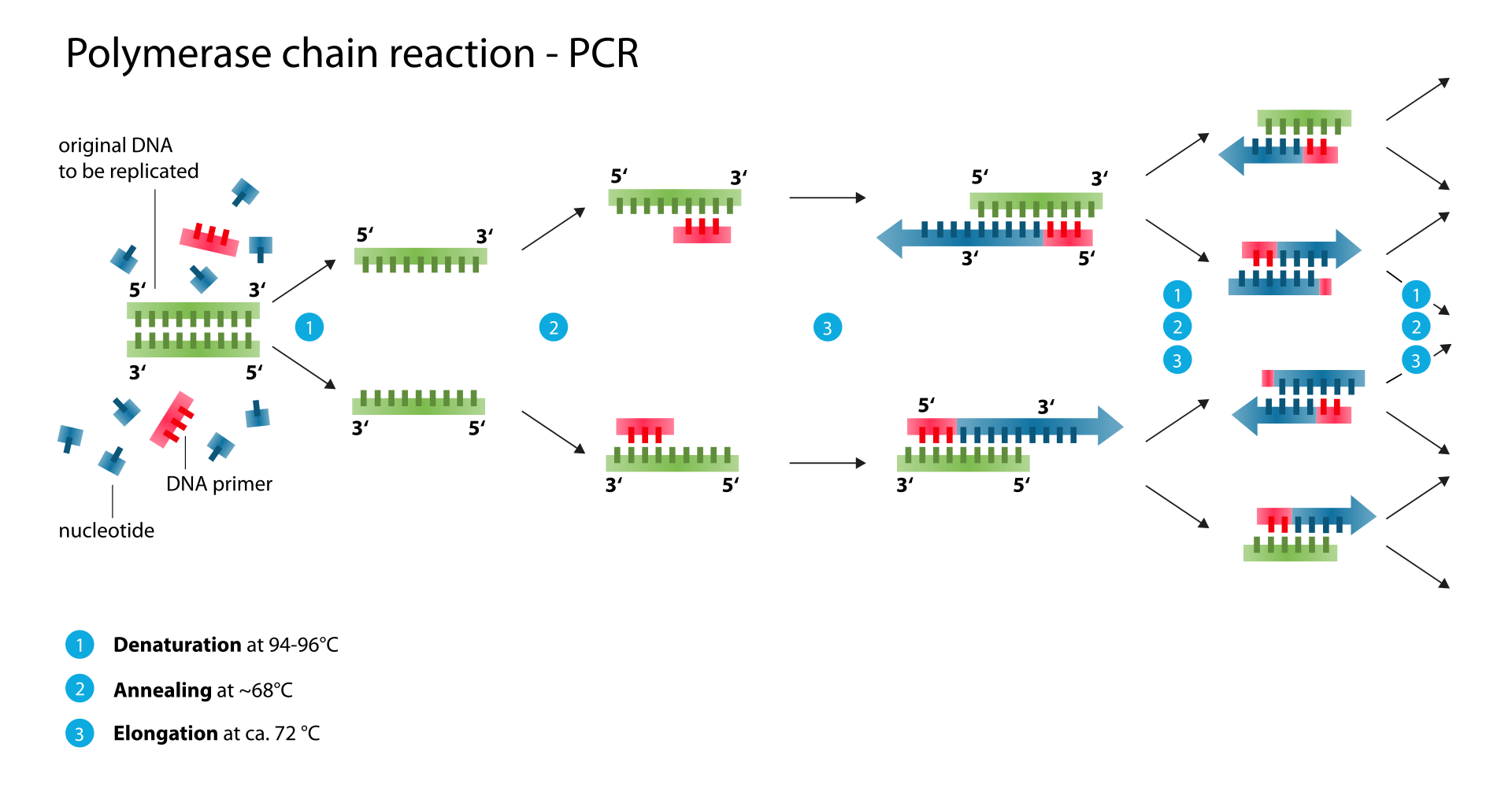
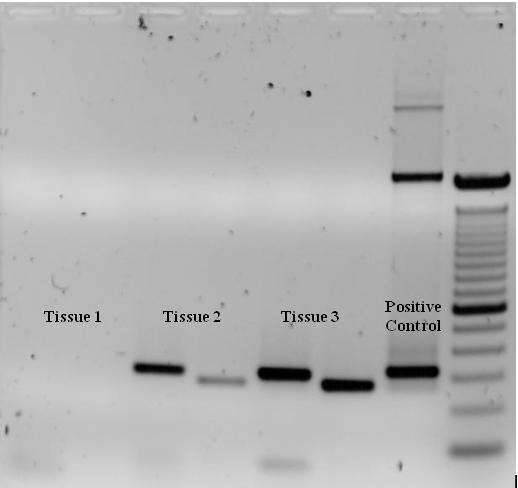
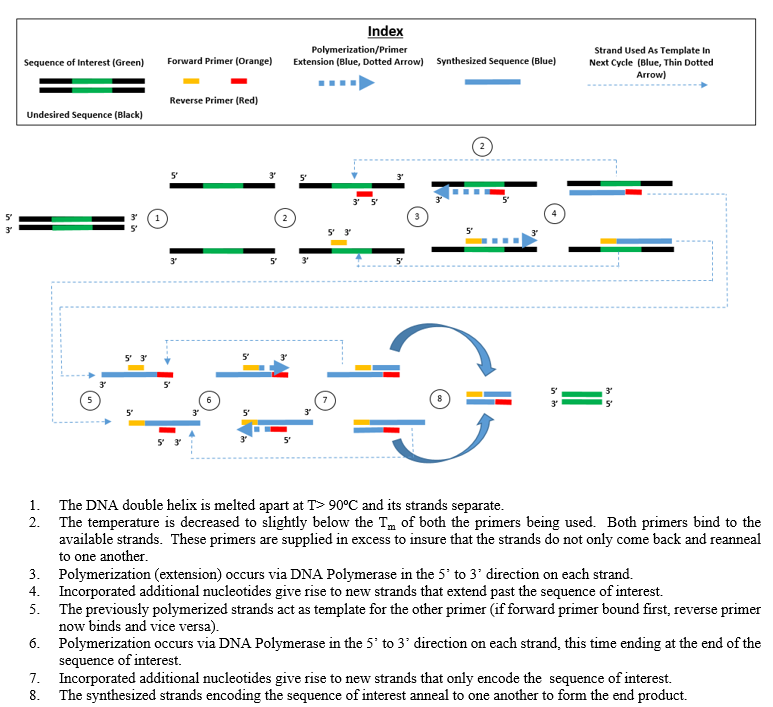
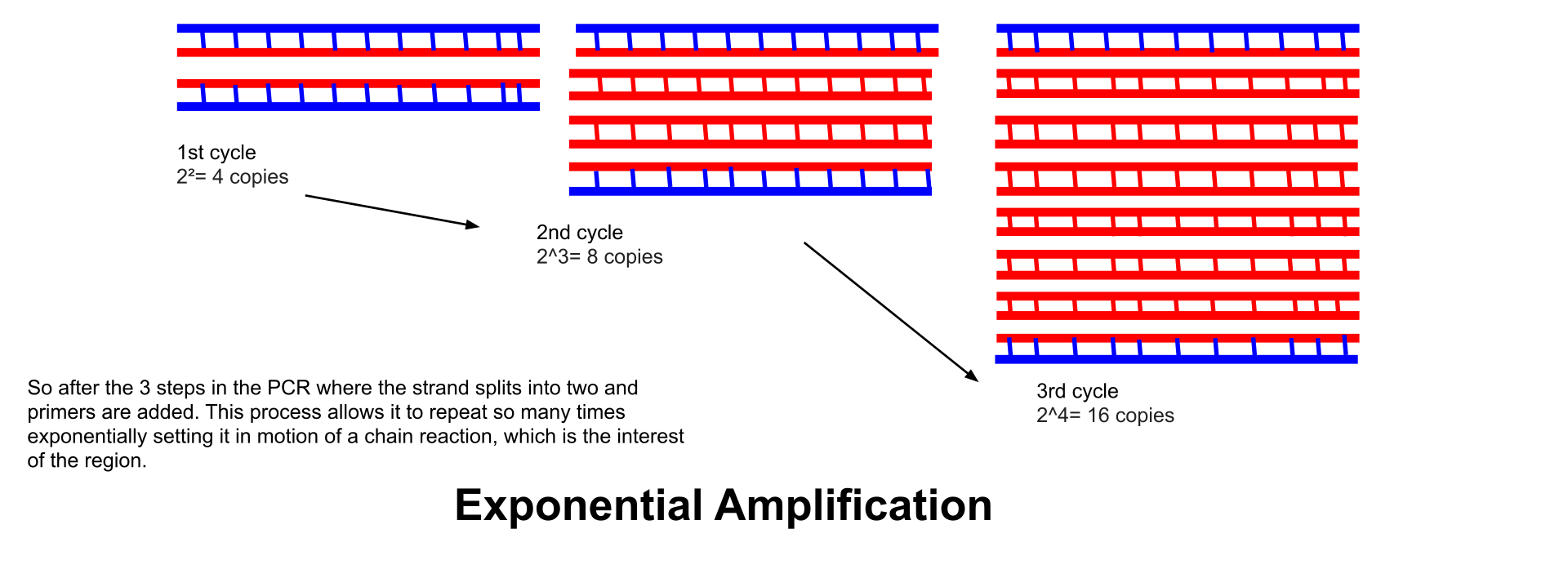
,_c_1986._(9663810586).jpg)